 7 minutes
7 minutes Ajouter un commentaire
Ajouter un commentaire
L’efficacité du molnupiravir est de l’ordre de 30 % pour réduire la progression de la COVID-19 vers une forme grave de l'infection (illustration).
Résumé
L'utilisation de l'antiviral LAGEVRIO 200 mg gélule (molnupiravir) en accès précoce n'a pas été autorisé par la Haute Autorité de Santé dans le traitement des formes légères à modérées de la maladie à coronavirus 2019 (COVID-19) chez les adultes ayant un test de diagnostic positif au SARS-CoV-2 et qui présentent au moins un facteur de risque de développer une forme sévère de la maladie.
L'attribution d'une autorisation en accès précoce (AAP) est basée sur différents critères, parmi lesquels la présomption d'innovation. Selon la HAS, LAGEVRIO ne répond pas à ce critère.
En outre, la décision de la HAS repose sur les données d'efficacité et de sécurité disponibles à ce jour (données issues de l'étude MOVe-OUT), lesquelles montrent une efficacité du molnupiravir d'environ 30 % pour réduire la progression vers une forme grave de l'infection. En comparaison, l'efficacité de la bithérapie d'anticorps monoclonaux RONAPREVE (casirivimab/imdevimab) est de l'ordre de 80 % sur ce même critère.
Enfin, les récentes dispositions réglementaires prévoyant un accès à LAGEVRIO en ville, la HAS pointe du doigt un "risque de perte de chance" pour des patients infectés et à risque de forme grave, chez lesquels RONAPREVE serait une meilleure option. Elle recommande d'ailleurs de faciliter l'accès aux anticorps monoclonaux pour le traitement curatif de la COVID, sur l'ensemble du territoire.
L'utilisation de l'antiviral LAGEVRIO 200 mg gélule (molnupiravir) en accès précoce n'a pas été autorisé par la Haute Autorité de Santé dans le traitement des formes légères à modérées de la maladie à coronavirus 2019 (COVID-19) chez les adultes ayant un test de diagnostic positif au SARS-CoV-2 et qui présentent au moins un facteur de risque de développer une forme sévère de la maladie.
L'attribution d'une autorisation en accès précoce (AAP) est basée sur différents critères, parmi lesquels la présomption d'innovation. Selon la HAS, LAGEVRIO ne répond pas à ce critère.
En outre, la décision de la HAS repose sur les données d'efficacité et de sécurité disponibles à ce jour (données issues de l'étude MOVe-OUT), lesquelles montrent une efficacité du molnupiravir d'environ 30 % pour réduire la progression vers une forme grave de l'infection. En comparaison, l'efficacité de la bithérapie d'anticorps monoclonaux RONAPREVE (casirivimab/imdevimab) est de l'ordre de 80 % sur ce même critère.
Enfin, les récentes dispositions réglementaires prévoyant un accès à LAGEVRIO en ville, la HAS pointe du doigt un "risque de perte de chance" pour des patients infectés et à risque de forme grave, chez lesquels RONAPREVE serait une meilleure option. Elle recommande d'ailleurs de faciliter l'accès aux anticorps monoclonaux pour le traitement curatif de la COVID, sur l'ensemble du territoire.
Dans un avis validé le 6 décembre 2021, la Haute Autorité de Santé a décidé de ne pas accorder d'autorisation d'accès précoce préalablement à l'AMM (cf. Infos pratiques VIDAL : le dispositif d'accès précoce) pour l'antiviral LAGEVRIO 200 mg gélule (molnupiravir), dans le traitement des formes légères à modérées de la maladie à coronavirus 2019 (COVID-19) chez les adultes ayant un test de diagnostic positif au SARS-CoV-2 et qui présentent au moins un facteur de risque de développer une forme sévère de la maladie [cf. Encadré 1].
Un suivi virologique rapproché est recommandé en cas d'échec thérapeutique.
Encadré 1 - Populations à risque élevé d'évolution vers une forme grave (population définie par l'ANRS-Maladies infectieuses)
|
Le molnupiravir, premier antiviral proposé pour lutter contre le SARS-CoV-2
Le molnupiravir (ou MK-4482) est un nouvel antiviral ciblant le SARS-CoV-2.
Il s'agit d'une prodrogue métabolisée en N-hydroxycytidine (NHC) analogue ribonucléosidique. Il devient pharmacologiquement actif (NHC-TP) après phosphorylation dans les cellules, et agit par inhibition de la réplication virale.
Le molnupiravir est le principe actif de la spécialité LAGEVRIO, présentée en gélule de 200 mg.
Une demande d'autorisation de mise sur le marché (AMM) conditionnelle a été déposée pour LAGEVRIO auprès de l'Agence européenne du médicament (EMA) en novembre 2021, dans une indication plus large que celle faisant l'objet de la demande d'accès précoce, à savoir "dans le traitement de la maladie à coronavirus 2019 (COVID-19) chez les adultes".
L'ANSM a pour sa part rendu un avis selon lequel l'efficacité et la sécurité de ce médicament étaient fortement présumées dans le traitement de la COVID-19 en cas d'impossibilité de recours aux anticorps monoclonaux chez les adultes ne nécessitant pas d'oxygénothérapie, et étant à risque élevé d'évolution vers une forme grave de la maladie, à savoir les populations suivantes telles que définies par l'ANRS-Maladies Infectieuses Émergentes (cf. Encadré 1 ci-dessus).
Efficacité de LAGEVRIO : les résultats de l'étude MOVe-OUT
L'efficacité et la tolérance du molnupiravir (800 mg 2 fois par jour) ont été principalement étudiées dans l'étude MOVe-OUT de phase IIb/III, randomisée, en double aveugle, contrôlée versus placebo, ayant prévue d'inclure 1 550 patients (environ 775 patients dans le groupe molnupiravir 800 mg et 775 patients dans le groupe placebo). Cette étude multicentrique a été réalisée dans 175 centres répartis dans 24 pays dont la France ayant inclus 7 patients.
Les patients inclus étaient atteints d'une forme légère à modérée de COVID-19, non hospitalisés, à risque de progression vers une forme grave de la maladie (âge > 60 ans, cancer évolutif, insuffisance rénale chronique, bronchopneumopathie chronique obstructive, obésité, antécédents cardiaques graves de type insuffisance cardiaque, maladie coronarienne ou cardiomyopathies, diabète).
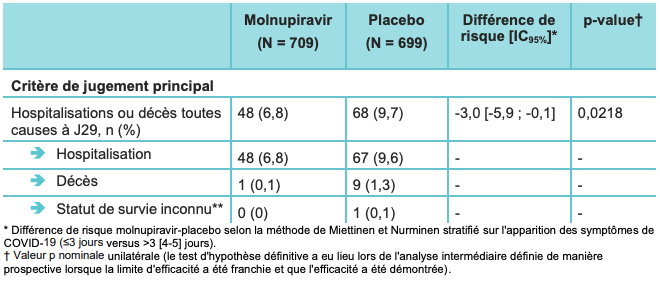
Le critère de jugement principal était le taux d'hospitalisation ou de décès, défini par la proportion de patients hospitalisés (séjour, ou soins intensifs, ou hospitalisation en réanimation) ou décédés à J29.
Selon les résultats de l'analyse intermédiaire (cf. Tableau I) :
- l'incidence des hospitalisations ou décès toutes causes à J29 (population ITTm) a été plus faible dans le groupe ayant reçu le molnupiravir que dans le groupe placebo : 6,8 % (48/709) versus 9,7 % (68/699) ;
- avec une différence ajustée de -3,0 %31 ; IC95% : [-5,9 ; -0,1] ; p = 0,0218 par rapport au placebo ;
- soit une réduction du risque relatif d'environ 30 % (IC95 % : [1% ; 51%]).
Tableau I - Principaux résultats de l'analyse finale de l'étude MOVe-OUT (population ITTm) [source : HAS - LAGEVRIO 200 mgGélule - Demande d'autorisation d'accès précoce - décembre 2021]
Après analyse des résultats, la HAS a souligné les points suivants :
- en termes de réduction des risques de progression vers une forme grave de COVID-19 : sur ce critère, la réduction du risque associée au molnupiravir est moindre (30 %) en comparaison à l'efficacité rapportée avec les anticorps monoclonaux casirivimab-imdevimab (RONAPREVE) évaluée à 80 % ;
- discordance importante entre l'analyse intermédiaire et l'analyse finale : dans l'analyse intermédiaire, la HAS note que l'efficacité du molnupiravir sur le risque de progression vers une forme grave est estimée à 50 %, alors que cette efficacité est de l'ordre de 30 % dans l'analyse finale ;
- molnupiravir et charge virale : la HAS regrette l'absence de données sur l'impact du molnupiravir sur la négativation de la charge virale (fait de réduire la présence du virus jusqu'à ce qu'il en devienne indétectable).
Conclusions de la HAS au regard du cadre défini pour l'autorisation en accès précoce
Sur la base des données d'efficacité et de tolérance disponibles, la HAS estime que LAGEVRIO n'est pas présumé innovant au regard des anticorps monoclonaux, du fait notamment des incertitudes majeures sur l'efficacité de ce traitement. Or, la présomption d'innovation est un critère à remplir pour bénéficier d'une autorisation d'accès précoce.
En termes de tolérance, le profil du molnupiravir apparaît favorable dans les études cliniques, sous réserve des risques identifiés dans le programme non clinique :
- risque fœtotoxique/tératogène, mutagène ;
- toxicité au niveau médullaire/hématologique, ainsi que des os et des cartilages.
LAGEVRIO en ville : la HAS pointe du doigt un risque de perte de chance
Conformément à un arrêté du 22 novembre publié au Journal officiel du 23 novembre 2021, la dispensation en officine des antiviraux par voie orale, dont le molnupiravir, indiqué dans la prise en charge de la COVID-19 et bénéficiant d'un accès précoce, est autorisée en pharmacie d'officine.
Ces dispositions réglementaires sont prises à titre dérogatoire, dans le cadre de l'état d'urgence sanitaire.
Pour la HAS, "l'accès à LAGEVRIO en ville risquerait d'induire une perte de chance pour les patients, qui ne se verraient pas traités par un traitement plus efficace, le RONAPREVE".
La HAS plaide pour un accès facilité au RONAPREVE
En parallèle de cette décision portant sur LAGEVRIO, la HAS encourage "l'accès facilité à RONAPREVE en curatif sur l'ensemble du territoire pendant la cinquième vague épidémique liée au variant delta" (cf. Encadré 2).
Encadré 2 - Indications de RONAPREVE dans le cadre de l'accès précoce
| Traitement de la COVID-19 : L'association casirivimab et imdevimab est indiquée dans le traitement de la COVID-19 confirmée par un test virologique de détection du SARS-CoV-2 positif, chez les patients âgés de 12 ans et plus :
Prévention de la COVID-19 : L'association casirivimab et imdevimab est indiquée :
Ou les patients séronégatifs après un schéma vaccinal complet ou non éligibles à la vaccination et qui présentent une immunodépression sévère et qui sont à haut risque de forme sévère de la COVID-19. |
Pour aller plus loin
Communiqué de presse - COVID-19 : deux nouveaux traitements évalués par la HAS (HAS, 10 décembre 2021)
Demande d'autorisation d'accès précoce pour LAGEVRIO 200 mg gélule (molnupiravir) (HAS, 6 décembre 2021)
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !





Commentaires
Cliquez ici pour revenir à l'accueil.