Mise à jour : 22 août 2024
Sommaire
Connectez-vous pour accéder à ce contenu
SYNTHÈSE |
Cip : 3400955089537
Modalités de conservation : Avant ouverture : -130° < durant 13 mois (Conserver au congélateur)
Après ouverture : 15° < t < 25° durant 2 heures (Ne pas recongeler)
FORMES et PRÉSENTATIONS |
BREYANZI 1,1 à 70 × 10 6 cellules/mL / 1,1 à 70 × 10 6 cellules/mL : dispersion pour perfusion (légèrement opaque à opaque, incolore à jaune ou brun-jaune).
Boîte contenant : boîtes individuelles de 1 à 4 flacon(s)* de chaque composant cellulaire CD8+ et CD4+, selon la concentration de lymphocytes T viables CAR-positifs dans chaque médicament cryopréservé.
* Flacon(s) de cryoconservation composé(s) de copolymère oléfine cyclique
Chaque flacon de 5 mL contient 4,6 mL de dispersion cellulaire.
COMPOSITION |
2.1 Description générale
Breyanzi (lisocabtagene maraleucel) est un produit anti-CD19 à base de cellules autologues génétiquement modifiées contenant des lymphocytes T CD8+ et CD4+ purifiés, de composition définie, transduits séparément ex vivo au moyen d'un vecteur lentiviral non réplicatif exprimant un récepteur antigénique chimérique (CAR) anti-CD19 comprenant un domaine de liaison du fragment variable monocaténaire [scFv]) dérivé d'un anticorps monoclonal murin dirigé spécifiquement contre le CD19 (Acm ; FMC63), une partie de l'endodomaine de costimulation 4-1BB, des domaines de signalisation de la chaîne zêta (ζ) du CD3 et un récepteur tronqué du facteur de croissance épidermique (EGFRt) non fonctionnel.
2.2 Composition qualitative et quantitative
Breyanzi contient des lymphocytes T viables CAR-positifs, avec une composition définie de composants cellulaires CD8+ et CD4+ :
Composant cellulaire CD8+
Chaque flacon contient du lisocabtagene maraleucel à une concentration dépendante du lot de cellules T autologues génétiquement modifiées pour exprimer un récepteur antigénique chimérique anti-CD19 (lymphocytes T viables CAR-positifs). Le médicament est conditionné dans un ou plusieurs flacon(s) contenant au total une dispersion cellulaire de 5,1 à 322 x 106 lymphocytes T viables CAR-positifs (1,1 à 70 x 106 lymphocytes T viables CAR-positifs/mL) en suspension dans une solution de cryoconservation.
Chaque flacon contient 4,6 mL de composant cellulaire CD8+.
Composant cellulaire CD4+
Chaque flacon contient du lisocabtagene maraleucel à une concentration dépendante du lot de cellules T autologues génétiquement modifiées pour exprimer un récepteur antigénique chimérique anti-CD19 (lymphocytes T viables CAR-positifs). Le médicament est conditionné dans un ou plusieurs flacon(s) contenant au total une dispersion cellulaire de 5,1 à 322 x 106 lymphocytes T viables CAR-positifs (1,1 à 70 x 106 lymphocytes T viables CAR-positifs/mL) en suspension dans une solution de cryoconservation.
Chaque flacon contient 4,6 mL de composant cellulaire CD4+.
Il est possible que plusieurs flacons de chaque composant cellulaire CD8+ et/ou CD4+ soient nécessaires pour obtenir la dose de Breyanzi. Le volume total à administrer et le nombre de flacons nécessaires peuvent être différents pour chaque composant cellulaire.
L'information quantitative concernant chaque composant cellulaire du médicament, y compris le nombre de flacons (voir rubrique Elimination/Manipulation) à administrer, est présentée dans le certificat de libération pour perfusion (RfIC) situé à l'intérieur du couvercle du conteneur d'expédition pour cryoconservation utilisé pour le transport. Le certificat RfIC pour chaque composant comporte le volume total à administrer, le nombre de flacons nécessaires et le volume à administrer de chaque flacon, en fonction de la concentration en lymphocytes T viables CAR-positifs cryopréservés.
Excipients à effet notoire
Ce médicament contient 12,5 mg de sodium, 6,5 mg de potassium et 0,35 mL (7,5 % v/v) de diméthylsulfoxyde (DMSO) par flacon (voir rubrique Mises en garde et précautions d'emploi).
Cryostor CS10, chlorure de sodium, gluconate de sodium, acétate de sodium trihydraté, chlorure de potassium, chlorure de magnésium, albumine humaine, N-acétyl-DL-tryptophane, acide caprylique, eau pour préparations injectables.
INDICATIONS |
Breyanzi est indiqué pour le traitement des patients adultes atteints d'un lymphome diffus à grandes cellules B (LDGCB), d'un lymphome B de haut grade (LBHG), d'un lymphome médiastinal primitif à grandes cellules B (LMPGCB) ou d'un lymphome folliculaire de grade 3B (LF3B) en rechute dans les 12 mois suivant la fin d'une immunochimiothérapie de première ligne ou réfractaire à ce traitement de première ligne.
Breyanzi est indiqué pour le traitement des patients adultes atteints d'un LDGCB, d'un LMPGCB ou d'un LF3B réfractaire ou en rechute après au moins deux lignes de traitement systémique.
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
CONTRE-INDICATIONS |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Traçabilité
Les exigences en matière de traçabilité des médicaments de thérapie innovante à base de cellules doivent s'appliquer. Afin de garantir la traçabilité, le nom du produit, le numéro de lot et le nom du patient traité doivent être conservés pendant une durée de 30 ans après l'expiration du produit.
Usage autologue
Breyanzi est destiné uniquement à un usage autologue et ne doit en aucun cas être administré à d'autres patients. Breyanzi ne doit pas être administré si les informations figurant sur les étiquettes du produit et sur le certificat de libération pour perfusion (RfIC) ne correspondent pas à l'identité du patient.
Raisons de retarder le traitement
En raison des risques associés au traitement par Breyanzi, la perfusion doit être reportée si un patient se trouve dans l'une des situations suivantes :
En cas de report de la perfusion de Breyanzi, voir la rubrique Posologie et mode d'administration.
Don de sang, d'organes, de tissus et de cellules
Les patients traités par Breyanzi ne doivent pas donner de sang, d'organes, de tissus ni de cellules à des fins de greffe.
Lymphome du système nerveux central (SNC)
Aucune donnée n'est disponible sur l'utilisation de Breyanzi chez les patients présentant un lymphome primitif du SNC. L'expérience clinique de l'utilisation de Breyanzi pour les lymphomes secondaires du SNC est limitée (voir rubrique Pharmacodynamie).
Administration antérieure d'une thérapie anti-CD19
L'expérience clinique de Breyanzi chez les patients ayant été exposés antérieurement à une thérapie anti-CD19 est limitée (voir rubrique Pharmacodynamie). Les données cliniques disponibles chez les patients ayant un statut CD19 négatif traités par Breyanzi sont limitées. Chez les patients ayant un statut CD19 négatif déterminé par immunohistochimie, l'expression du CD19 est néanmoins possible. Les bénéfices et risques potentiels associés au traitement par Breyanzi chez les patients ayant un statut CD19 négatif doivent être évalués.
Syndrome de relargage des cytokines
Des cas de SRC, y compris des réactions fatales ou engageant le pronostic vital, peuvent survenir après la perfusion de Breyanzi. Chez les patients ayant reçu préalablement une ligne de traitement pour un lymphome à grandes cellules B (LGCB), le délai médian d'apparition du SRC était de 4 jours (intervalle : 1 à 63 jours, la limite supérieure étant due à l'apparition d'un SRC sans fièvre rapporté chez un patient). Chez les patients ayant reçu préalablement au moins deux lignes de traitement pour un LGCB, le délai médian d'apparition du SRC était de 4 jours (intervalle : 1 à 14 jours). Moins de la moitié des patients traités par Breyanzi ont présenté, à des degrés divers, un SRC (voir rubrique Effets indésirables).
Dans les études cliniques, une charge tumorale élevée présente avant la perfusion de Breyanzi a été associée à une incidence plus élevée de SRC.
Le tocilizumab et/ou un corticoïde ont été utilisés pour prendre en charge le SRC après la perfusion de Breyanzi (voir rubrique Effets indésirables).
Surveillance et prise en charge du SRC
Le SRC doit être identifié à partir des manifestations cliniques. Les patients doivent être évalués et traités pour les autres causes de fièvre, d'hypoxie et d'hypotension artérielle.
Avant la perfusion de Breyanzi, au moins une dose de tocilizumab par patient doit être disponible sur place. Le centre de traitement doit avoir accès à une dose supplémentaire de tocilizumab dans les 8 heures suivant l'administration de chaque dose précédente. Dans le cas exceptionnel où le tocilizumab ne serait pas disponible en raison d'une pénurie figurant dans la liste des pénuries de l'Agence européenne des médicaments, le centre de traitement doit avoir accès à des alternatives appropriées pour prendre en charge un SRC en remplacement du tocilizumab. Les signes et symptômes de SRC doivent être surveillés chez les patients à 2 ou 3 reprises pendant la première semaine suivant la perfusion de Breyanzi dans l'établissement de santé qualifié. Après la première semaine, la fréquence de surveillance du patient sera laissée à la discrétion du médecin, et sera maintenue pendant au moins 4 semaines après la perfusion. Il doit être conseillé aux patients de consulter un médecin immédiatement en cas d'apparition, à n'importe quel moment, de signes ou symptômes du SRC afin d'être pris en charge sans délai.
Aux premiers signes de SRC, un traitement par soins de support, par tocilizumab ou tocilizumab associé aux corticoïdes doit être instauré, comme indiqué dans le tableau 1. Breyanzi poursuit son expansion après l'administration de tocilizumab et de corticoïdes (voir rubrique Pharmacocinétique).
La fonction cardiaque et la fonction des autres organes des patients qui présentent un SRC doivent être étroitement surveillées jusqu'à la résolution des symptômes. En cas de SRC sévère ou engageant le pronostic vital, l'admission en unité de soins intensifs pour surveillance et administration de soins de support doit être envisagée.
Des investigations pour rechercher une lymphohistiocytose hémophagocytaire/un syndrome d'activation macrophagique (LHH/SAM) doivent être envisagées chez les patients présentant un SRC sévère ou ne répondant pas aux traitements. Le traitement de la LHH/du SAM doit être administré conformément aux recommandations de l'établissement.
Si une toxicité neurologique concomitante est suspectée pendant un SRC, administrer :
Grade du SRCa |
Tocilizumab |
Corticoïdesb |
Grade 1 |
En cas d'apparition 72 heures ou plus après la perfusion, traiter de façon symptomatique. |
En cas d'apparition 72 heures ou plus après la perfusion, traiter de façon symptomatique. |
Grade 2 |
Administrer du tocilizumab à la dose de 8 mg/kg IV sur 1 heure (sans dépasser 800 mg). |
En cas d'apparition 72 heures ou plus après la perfusion, envisager l'administration de 10 mg IV de dexaméthasone toutes les 12 à 24 heures. |
En l'absence d'amélioration dans les 24 heures ou en cas de progression rapide, répéter l'administration de tocilizumab et augmenter la dose et la fréquence de la dexaméthasone (10 à 20 mg IV toutes les 6 à 12 heures). |
||
Grade 3 |
Identique au grade 2. |
Administrer 10 mg de dexaméthasone IV toutes les 12 heures. |
En l'absence d'amélioration dans les 24 heures ou en cas de progression rapide du SRC, augmenter le tocilizumab et les corticoïdes conformément au grade 2. |
||
Grade 4 |
Identique au grade 2. |
Administrer 20 mg de dexaméthasone IV toutes les 6 heures. |
En l'absence d'amélioration dans les 24 heures ou en cas de progression rapide du SRC, augmenter le tocilizumab et les corticoïdes conformément au grade 2. |
||
a Lee et al. 2014.
b Si les corticoïdes sont instaurés, continuer pendant au moins 3 doses ou jusqu'à disparition complète des symptômes avant d'envisager de réduire progressivement la dose.
Effets indésirables neurologiques
Des toxicités neurologiques, incluant le syndrome de neurotoxicité lié aux cellules effectrices immunitaires (ICANS), pouvant être fatales ou engager le pronostic vital, sont survenues après le traitement par Breyanzi, y compris de façon concomitante à un SRC, après résolution d'un SRC ou en l'absence de SRC. Le délai médian d'apparition du premier événement était de 8 jours (intervalle : 1 à 63 jours) chez les patients ayant reçu préalablement une ligne de traitement pour un LGCB et de 9 jours (intervalle : 1 à 66 jours) chez les patients ayant reçu préalablement au moins deux lignes de traitement. Les symptômes neurologiques les plus fréquents étaient l'encéphalopathie, les tremblements, l'aphasie, le délire, les vertiges et les céphalées (voir rubrique Effets indésirables).
Surveillance et prise en charge des toxicités neurologiques
Les signes et symptômes de toxicités neurologiques doivent être surveillés chez les patients à 2 ou 3 reprises pendant la première semaine suivant la perfusion dans l'établissement de santé qualifié. Après la première semaine, la fréquence de surveillance du patient sera définie à la discrétion du médecin, et sera maintenue pendant au moins 4 semaines après la perfusion. Il doit être conseillé aux patients de consulter un médecin immédiatement en cas d'apparition, à n'importe quel moment, de signes et symptômes de toxicités neurologiques afin d'être pris en charge sans délai.
Si une toxicité neurologique est suspectée, elle doit être prise en charge conformément aux recommandations du tableau 2. Les autres causes de symptômes neurologiques, incluant les événements vasculaires, doivent être écartées. Des soins de support dispensés en unité de soins intensifs doivent être administrés en cas de toxicités neurologiques sévères ou engageant le pronostic vital.
Si un SRC concomitant est suspecté pendant la toxicité neurologique, administrer :
Grade de toxicité neurologique et présentation des symptômesa |
Corticoïdes et anticonvulsivants |
Grade 1* |
Commencer les anticonvulsivants non sédatifs (par exemple, le lévétiracétam) en prévention des convulsions. |
Si 72 heures ou plus après la perfusion, surveiller. |
|
Si moins de 72 heures après la perfusion, administrer 10 mg de dexaméthasone en IV toutes les 12 à 24 heures pendant 2 à 3 jours. |
|
Grade 2* |
Commencer les anticonvulsivants non sédatifs (par exemple, le lévétiracétam) en prévention des convulsions. |
Administrer la dexaméthasone 10 mg IV toutes les 12 heures pendant 2 à 3 jours ou plus en cas de symptômes persistants. Envisager une réduction progressive en cas d'exposition cumulée aux corticoïdes supérieure à 3 jours. |
|
En l'absence d'amélioration après 24 heures ou en cas d'aggravation de la toxicité neurologique, augmenter la dose et/ou la fréquence de la dexaméthasone jusqu'à un maximum de 20 mg IV toutes les 6 heures. |
|
En l'absence d'amélioration après 24 heures supplémentaires, en cas de progression rapide des symptômes ou de complications engageant le pronostic vital, administrer de la méthylprednisolone (dose de charge de 2 mg/kg, suivie de 2 mg/kg répartis en 4 administrations par jour ; diminuer progressivement sur 7 jours). |
|
Grade 3*
Ou élévation de la PICc : œdème focal/local visible sur les examens de neuro-imagerie. |
Commencer les anticonvulsivants non sédatifs (par exemple, le lévétiracétam) en prévention des convulsions. |
Administrer de la dexaméthasone 10 à 20 mg IV toutes les 8 à 12 heures. Les corticoïdes ne sont pas recommandés pour des céphalées isolées de grade 3. |
|
En l'absence d'amélioration après 24 heures ou en cas d'aggravation de la toxicité neurologique, passer à la méthylprednisolone (dose et fréquence identiques au grade 2). |
|
Si un œdème cérébral est suspecté, envisager une hyperventilation et un traitement hyperosmolaire. Administrer de la méthylprednisolone à haute dose (1 à 2 g, à renouveler toutes les 24 heures si nécessaire ; diminuer progressivement selon la situation clinique) et du cyclophosphamide à la dose de 1,5 g/m2. |
|
Grade 4*
Ou convulsionsc soit :
Ou troubles moteursc :
Ou élévation de la PIC/œdème cérébralc accompagné(e) de signes/symptômes tels que :
|
Commencer les anticonvulsivants non sédatifs (par exemple, le lévétiracétam) en prévention des convulsions. |
Administrer de la dexaméthasone 20 mg IV toutes les 6 heures. |
|
En l'absence d'amélioration après 24 heures ou en cas d'aggravation de la toxicité neurologique, passer à la méthylprednisolone (dose et fréquence identiques au grade 2). |
|
Si un œdème cérébral est suspecté, envisager une hyperventilation et un traitement hyperosmolaire. Administrer de la méthylprednisolone à haute dose (1 à 2 g, à renouveler toutes les 24 heures si nécessaire ; diminuer progressivement selon la situation clinique) et du cyclophosphamide à la dose de 1,5 g/m2. |
EEG = électroencéphalogramme ; ICE = encéphalopathie liée aux cellules effectrices immunitaires (Immune Effector Cell-Associated Encephalopathy) ; PIC = pression intracrânienne
* Détermination du grade d'après les critères NCI CTCAE ou ASTCT/ICANS
a La prise en charge est déterminée par l'événement le plus sévère non attribuable à une autre cause.
b Si le patient est éveillable et est capable de se soumettre à une évaluation de l'ICE, évaluer : l'orientation (sait indiquer l'année, le mois, la ville et l'établissement hospitalier = 4 points), la capacité à nommer (lui demander de nommer 3 objets, p. ex., lui montrer une horloge, un crayon et un bouton = 3 points), la capacité à suivre des instructions (p. ex., « montrez-moi 2 doigts » ou « fermez les yeux et tirez la langue » = 1 point), l'écriture (capacité à écrire une phrase basique = 1 point) et l'attention (compter à l'envers de 10 en 10, en partant de 100 = 1 point). Si le patient ne peut pas être réveillé et n'est pas capable de se soumettre à une évaluation de l'ICE (ICANS de grade 4) = 0 point.
c Attribuable à aucune autre cause.
Infections et neutropénie fébrile
Breyanzi ne doit pas être administré aux patients présentant une infection active cliniquement significative ou un trouble inflammatoire. Des infections sévères, y compris des infections ayant engagé le pronostic vital ou ayant été fatales, ont été observées chez des patients après la perfusion de ce médicament (voir rubrique Effets indésirables). Les signes et symptômes d'infection doivent être recherchés chez les patients avant et après l'administration et doivent être traités de manière appropriée. Des traitements antimicrobiens à visée prophylactique doivent être administrés conformément aux lignes directrices de l'établissement.
Une neutropénie fébrile a été observée chez des patients après le traitement par Breyanzi (voir rubrique Effets indésirables) et peut survenir de manière concomitante avec un SRC. En cas de neutropénie fébrile, l'infection doit être évaluée et traitée par des antibiotiques à large spectre, des solutés de remplissage et d'autres soins de support selon les indications médicales.
Les patients traités par Breyanzi peuvent présenter un risque accru d'infections COVID-19 sévères ou d'issue fatale. Les patients doivent être informés de l'importance des mesures de prévention.
Réactivation virale
Une réactivation virale (par exemple VHB, herpèsvirus humain de type 6 [HHV-6]) peut se produire chez les patients immunodéprimés.
Les manifestations de la réactivation virale peuvent compliquer et retarder le diagnostic et le traitement approprié des événements indésirables liés aux lymphocytes CAR-T. Des examens diagnostiques appropriés doivent être réalisés afin d'aider à distinguer ces manifestations des événements indésirables liés aux lymphocytes CAR-T.
Une réactivation du VHB, entraînant dans certains cas une hépatite fulminante, une insuffisance hépatique et le décès, peut se produire chez les patients traités par des médicaments dirigés contre les lymphocytes B. Chez les patients ayant des antécédents d'infection par le VHB, un traitement prophylactique antiviral suppressif est recommandé pour prévenir la réactivation du VHB pendant et après le traitement par Breyanzi (voir rubrique Pharmacodynamie).
Tests sérologiques
Un dépistage du VHB, du VHC et du VIH doit être réalisé avant le prélèvement des cellules destinées à la production (voir rubrique Posologie et mode d'administration).
Cytopénies prolongées
Les patients peuvent présenter des cytopénies pendant plusieurs semaines après la chimiothérapie lymphodéplétive et le traitement par Breyanzi (voir rubrique Effets indésirables). Une numération sanguine doit être contrôlée avant et après l'administration de Breyanzi. Les cytopénies prolongées doivent être traitées conformément aux recommandations cliniques.
Hypogammaglobulinémie
Une aplasie des lymphocytes B entraînant une hypogammaglobulinémie peut survenir chez les patients recevant un traitement par Breyanzi. L'hypogammaglobulinémie est très fréquemment observée chez les patients traités par Breyanzi (voir rubrique Effets indésirables). Les taux d'immunoglobulines doivent faire l'objet d'une surveillance après le traitement et toute diminution doit être prise en charge conformément aux recommandations cliniques, qui incluent des précautions pour prévenir les infections, une prophylaxie antibiotique et/ou un traitement substitutif par immunoglobulines.
Tumeurs malignes secondaires
Les patients traités par Breyanzi peuvent développer des tumeurs malignes secondaires. Les tumeurs malignes secondaires doivent être recherchées chez les patients tout au long de leur vie. En cas de survenue d'une tumeur maligne secondaire issue des lymphocytes T, il convient de contacter le laboratoire pharmaceutique afin d'obtenir des instructions sur le prélèvement des échantillons tumoraux pour analyse.
Syndrome de lyse tumorale (SLT)
Un SLT peut survenir chez les patients traités par des thérapies CAR-T. Pour limiter le risque de SLT, de l'allopurinol ou un autre traitement prophylactique doit être administré avant la perfusion de Breyanzi aux patients présentant un taux d'acide urique élevé ou une charge tumorale élevée. Les signes et symptômes de SLT doivent être surveillés et pris en charge conformément aux recommandations cliniques.
Réactions d'hypersensibilité
Des réactions allergiques peuvent se produire avec la perfusion de Breyanzi. De graves réactions d'hypersensibilité, y compris une anaphylaxie, peuvent être dues au diméthylsulfoxyde (DMSO).
Transmission d'un agent infectieux
Bien que Breyanzi soit soumis à des tests de stérilité et de recherche de mycoplasmes, il existe un risque de transmission d'agents infectieux. Les professionnels de santé qui administrent Breyanzi doivent par conséquent surveiller les patients pour détecter tout signe ou symptôme d'infection après le traitement et les traiter de façon adéquate, si nécessaire.
Interférence avec les tests virologiques
En raison de la présence de courtes séquences génétiques dans le vecteur lentiviral utilisé pour créer Breyanzi, identiques à des séquences génétiques du VIH, certains tests de détection d'acide nucléique du VIH (NAT) pourraient montrer un résultat positif sans lien avec une infection par le VIH.
Antécédents de greffe de cellules souches
L'administration du traitement aux patients ayant reçu une greffe allogénique de cellules souches et qui présentent une GvH aiguë ou chronique active n'est pas recommandée en raison du risque potentiel d'aggravation de la GvH par Breyanzi.
Suivi à long terme
Les patients doivent être inclus dans un registre et feront l'objet d'un suivi à partir de ce registre, ce qui permettra de mieux comprendre la sécurité et l'efficacité à long terme de Breyanzi.
Excipients
Ce médicament contient 12,5 mg de sodium par flacon, ce qui équivaut à 0,6 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
Ce médicament contient 0,2 mmol (ou 6,5 mg) de potassium par flacon. À prendre en compte chez les patients insuffisants rénaux ou chez les patients contrôlant leur apport alimentaire en potassium.
INTERACTIONS |
Connectez-vous pour accéder à ce contenu
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Chez les femmes en âge de procréer, un test de grossesse doit être réalisé avant le début du traitement par Breyanzi.
Se reporter aux informations concernant les modalités de prescription du cyclophosphamide et de la fludarabine pour en savoir plus sur la nécessité d'une contraception efficace chez les patients recevant une chimiothérapie lymphodéplétive.
Les données d'exposition sont insuffisantes pour permettre une recommandation sur la durée de contraception après un traitement par Breyanzi.
Grossesse
Il n'existe pas de données sur l'utilisation du lisocabtagene maraleucel chez la femme enceinte. Il n'a pas été conduit d'études de toxicité sur la reproduction et le développement chez l'animal permettant d'évaluer la survenue d'effets délétères chez le fœtus lorsqu'il est administré pendant la grossesse (voir rubrique Sécurité préclinique).
Le risque de transmission du lisocabtagene maraleucel au fœtus n'est pas connu. En se basant sur le mécanisme d'action, si les cellules transduites traversent le placenta, elles peuvent causer une toxicité fœtale, notamment une lymphopénie à lymphocytes B. Par conséquent, Breyanzi n'est pas recommandé pendant la grossesse ni chez les femmes en âge de procréer n'utilisant pas de contraception. Les femmes enceintes doivent être informées des risques pour le fœtus. Une grossesse après traitement par Breyanzi doit être discutée avec le médecin traitant.
L'évaluation des taux d'immunoglobulines et de lymphocytes B chez les nouveau-nés dont la mère a été traitée par Breyanzi doit être envisagée.
Allaitement
L'excrétion du lisocabtagene maraleucel dans le lait maternel ou sa transmission à l'enfant allaité n'est pas connue. Les femmes qui allaitent doivent être averties du risque potentiel pour l'enfant allaité.
Fertilité
Aucune donnée n'est disponible sur l'effet du lisocabtagene maraleucel sur la fertilité.
CONDUITE et UTILISATION DE MACHINES |
Breyanzi peut avoir une influence importante sur l'aptitude à conduire des véhicules et à utiliser des machines.
En raison des événements neurologiques potentiels, incluant une altération de l'état mental ou des crises convulsives sous Breyanzi, les patients recevant Breyanzi doivent éviter de conduire des véhicules ou d'utiliser des machines lourdes ou potentiellement dangereuses pendant au moins 8 semaines après la perfusion de Breyanzi.
EFFETS INDÉSIRABLES |
Connectez-vous pour accéder à ce contenu
SURDOSAGE |
Aucune donnée clinique n'est disponible concernant le surdosage de Breyanzi.
PHARMACODYNAMIE |
Connectez-vous pour accéder à ce contenu
PHARMACOCINÉTIQUE |
Connectez-vous pour accéder à ce contenu
SÉCURITÉ PRÉCLINIQUE |
Aucune étude de génotoxicité ou de cancérogenèse n'a été réalisée avec Breyanzi.
Les études d'expansion in vitro menées chez des donneurs sains et des patients n'ont révélé aucun signe de transformation et/ou d'immortalisation, ni d'intégration préférentielle près des gènes concernés dans les lymphocytes T de Breyanzi.
Compte tenu de la nature du produit, il n'a pas été conduit d'études non cliniques sur la fertilité, la reproduction et le développement.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
DURÉE DE CONSERVATION |
Flacon non ouvert stocké dans la phase vapeur de l'azote liquide
13 mois.
Après décongélation
Le produit doit être administré immédiatement après décongélation. La durée de conservation après décongélation ne doit pas dépasser 2 heures à température ambiante (15 °C à 25 °C).
Ne pas recongeler.
PRÉCAUTIONS PARTICULIÈRES DE CONSERVATION |
Breyanzi doit être conservé et transporté congelé dans la phase vapeur de l'azote liquide (≤ -130 °C) et être maintenu congelé jusqu'à ce que le patient soit prêt à recevoir le traitement afin de garantir que des cellules viables sont disponibles pour l'administration au patient. Le médicament décongelé ne doit pas être recongelé.
Pour les conditions de conservation du médicament après décongélation, voir la rubrique Durée de conservation.
PRÉCAUTIONS PARTICULIÈRES D'ÉLIMINATION ET DE MANIPULATION |
Précautions à prendre avant la manipulation ou l'administration du médicament
Préparation avant l'administration
Avant la décongélation des flacons
Décongélation des flacons
Préparation de la dose
Le prélèvement du volume de cellules requis de chaque flacon dans une seringue distincte doit être réalisé en suivant les instructions ci-après :
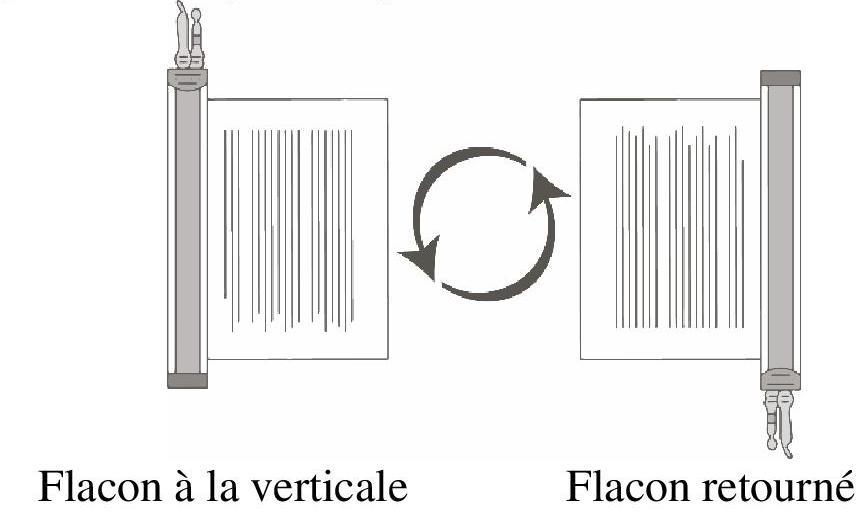 |
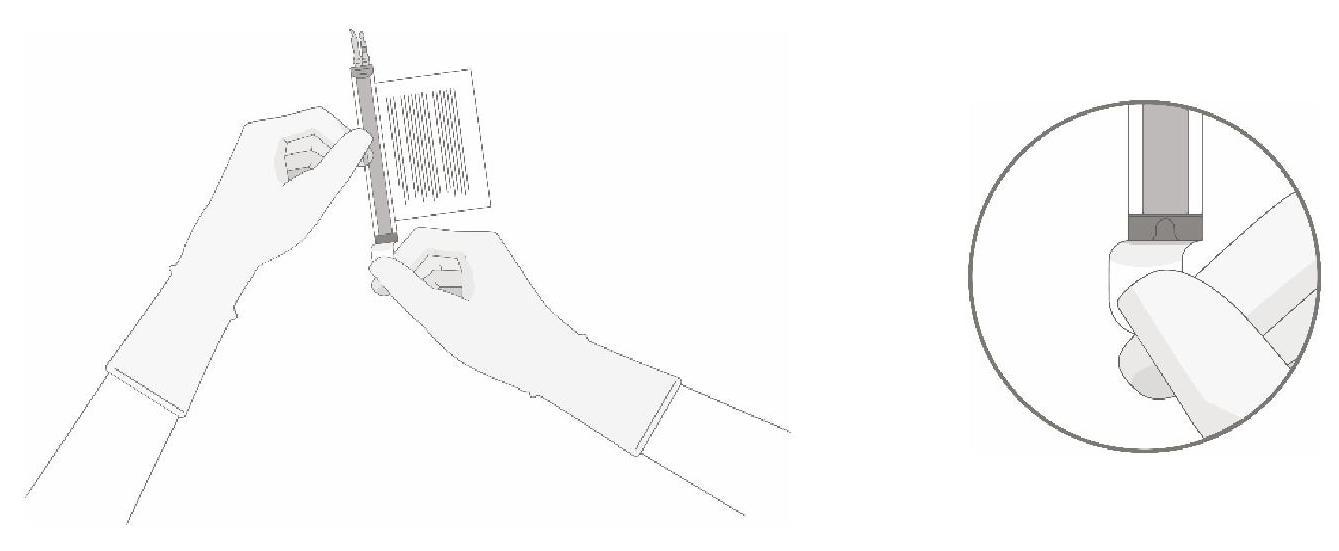

 |
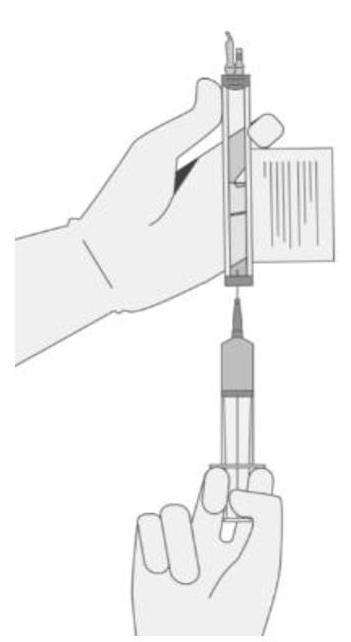 |

Administration
Pour des informations supplémentaires sur l'administration, voir rubrique Posologie et mode d'administration.
Mesures à prendre en cas d'exposition accidentelle
Précautions à prendre pour l'élimination du médicament
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM |
|
Agréé Collect et inscrit sur la liste des spécialités prises en charge en sus des GHS dans l’indication « traitement des patients adultes atteints d'un lymphome diffus à grandes cellules B (LDGCB), d'un lymphome B de haut grade (LHGCB), d'un lymphome médiastinal primitif à grandes cellules B (LMPGCB) ou d'un lymphome folliculaire de grade 3B (LF3B) en rechute dans les 12 mois suivant la fin d'une immunochimiothérapie de première ligne ou réfractaire à ce traitement de première ligne ».
En outre, l'inscription de la spécialité Breyanzi sur la liste des spécialités pharmaceutiques agréées à l'usage des collectivités et divers services publics est limitée à un nombre restreint de centres qualifiés à l'usage des médicaments à base de cellules CAR-T compte tenu de la complexité de la procédure. Dans ce contexte, la commission de la transparence souligne l'importance d'une prise en charge globale (incluant notamment les déplacements et les hébergements à proximité des établissements de santé qualifiés, lorsque nécessaire).
Prix et tarif de responsabilité (HT) par UCD :
UCD 3400890024518 (flacon) : 345000,000 euros.
Non agréé Collect dans l’indication « traitement des patients adultes atteints d'un LDGCB, d'un LMPGCB ou d'un LF3B réfractaire ou en rechute après au moins deux lignes de traitement systémique ».
Titulaire de l'AMM : Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Irlande.
