Classification pharmacothérapeutique VIDAL
Cancérologie - Hématologie > Antinéoplasiques > Chimiothérapie cytotoxique > Agents du fuseau (Stabilisants du fuseau : taxanes)
Urologie - Néphrologie > Cancer de la prostate (Stabilisants du fuseau : taxanes)
Classification ATC
ANTINEOPLASIQUES ET IMMUNOMODULATEURS > ANTINEOPLASIQUES > ALCALOÏDES VEGETAUX ET AUTRES MEDICAMENTS D'ORIGINE NATURELLE > TAXANES (CABAZITAXEL)
Excipients
polysorbate 80,
acide citrique excipient du solvant : eau ppi
Excipients à effet notoire :
EEN sans dose seuil : éthanol à 96 %
Présentation
JEVTANA 60 mg S à diluer et solvant p perf Fl/1,5ml+Fl solv/4,5ml
Cip : 3400957984977
Modalités de conservation : Avant ouverture : < 30° durant 36 mois (Ne pas conserver au réfrigérateur)
| Solution à diluer : | p flacon* |
Cabazitaxel
| 60 mg |
Excipients : polysorbate 80, acide citrique.
Solvant : éthanol à 96 %, eau pour préparations injectables.
Un ml de solution à diluer contient 40 mg de cabazitaxel.
Après dilution initiale avec la totalité du solvant, chaque ml de la solution contient 10 mg de cabazitaxel.
*
Le flacon de solution à diluer de 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,67 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation. Ce surremplissage permet d'obtenir une solution contenant 10 mg/ml de cabazitaxel (au moins 6 ml de volume extractible) après dilution de la solution à diluer avec la
totalité du contenu du flacon de solvant fourni.
Excipient à effet notoire : éthanol à 96 % (573,3 mg/flacon de solvant).
Jevtana en association à la prednisone ou la prednisolone est indiqué dans le traitement des patients adultes avec un cancer de la prostate métastatique résistant à la castration précédemment traités par un traitement à base de docétaxel (
cf Pharmacodynamie).
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
-
Réactions d'hypersensibilité :
- Tous les patients doivent recevoir une prémédication avant l'initiation de la perfusion de cabazitaxel (cf Posologie et Mode d'administration).
- Les patients doivent être étroitement surveillés pour les réactions d'hypersensibilité, essentiellement pendant la première et la seconde perfusion. Les réactions d'hypersensibilité peuvent survenir dans les quelques minutes suivant l'initiation de la perfusion de cabazitaxel, ainsi les installations et équipements pour le traitement de l'hypotension et de bronchospasmes devraient être à proximité du patient. Des réactions sévères peuvent survenir, incluant rash/érythèmes généralisés, hypotension et bronchospasmes. Les réactions d'hypersensibilité sévères nécessitent un arrêt immédiat du cabazitaxel et un traitement approprié. Les patients avec une réaction d'hypersensibilité doivent arrêter le traitement par Jevtana (cf Contre-indications).
-
Myélosuppression :
- Une myélosuppression se manifestant par une neutropénie, anémie, thrombopénie ou pancytopénie (voir « Risque de neutropénies » et « Anémie » en rubrique Mises en garde et Précautions d'emploi ci-dessous) peut survenir.
-
Risque de neutropénies :
- Les patients traités par cabazitaxel peuvent recevoir une prophylaxie par G-CSF, conformément aux guidelines de l'American Society of Clinical Oncology (ASCO) et/ou aux recommandations institutionnelles en vigueur, pour réduire le risque ou prendre en charge les complications des neutropénies (neutropénies fébriles, neutropénies prolongées ou infections neutropéniques).
- Une prophylaxie primaire avec G-CSF doit être considérée chez les patients ayant des facteurs de risque clinique important (âge > 65 ans, mauvais état général, épisodes précédents de neutropénie fébrile, champ d'irradiation antérieur extensif, mauvais état nutritionnel, ou d'autres facteurs de comorbidités sévères) qui les prédisposent à une augmentation des complications liées à une neutropénie prolongée.
- L'utilisation de G-CSF a montré qu'elle limitait l'incidence et la sévérité des neutropénies.
- La neutropénie est l'effet indésirable le plus fréquent du cabazitaxel (cf Effets indésirables). Le suivi de la numération formule sanguine hebdomadaire est essentiel pendant le cycle 1 et ensuite avant chaque cycle de traitement, afin d'ajuster la dose si besoin.
- La dose doit être réduite en cas de neutropénie fébrile, ou neutropénie prolongée malgré un traitement approprié (cf Posologie et Mode d'administration).
- Le traitement ne devra être repris chez ces patients seulement quand les neutrophiles seront ≥ 1500/mm3 (cf Contre-indications).
-
Affections gastro-intestinales :
- Des symptômes tels que des douleurs et une sensibilité abdominales, fièvre, constipation persistante, diarrhée, avec ou sans neutropénie, peuvent être des manifestations précoces d'une toxicité gastro-intestinale et doivent être évalués et traités rapidement. Le traitement par cabazitaxel peut alors être repoussé ou arrêté si nécessaire.
-
Risque de nausée, vomissement, diarrhée et déshydratation :
- Si des patients ont eu des antécédents de diarrhées après une administration de cabazitaxel, ils doivent être traités par des médicaments antidiarrhéiques habituellement utilisés. Des mesures appropriées doivent être prises pour réhydrater ces patients. Des diarrhées peuvent se produire plus fréquemment chez des patients ayant reçu une irradiation abdomino-pelvienne. Une déshydratation est plus fréquente chez les patients âgés de 65 ans et plus. Des mesures appropriées doivent être prises pour réhydrater les patients, les suivre et corriger leur taux sérique d'électrolytes, notamment le potassium. Un report du traitement ou une réduction de la dose peuvent être nécessaires pour des diarrhées de grade ≥ 3 (cf Posologie et Mode d'administration). Si des patients ont eu des antécédents de nausées et vomissements, ils doivent être traités avec des antiémétiques habituellement utilisés.
-
Risque de réactions gastro-intestinales graves :
- Des hémorragies et des perforations digestives, des iléus, des colites, incluant des issues fatales ont été rapportées chez des patients traités par cabazitaxel (cf Effets indésirables). Une attention est requise chez les patients les plus à risque de développer des complications digestives : ceux souffrant de neutropénie, les patients âgés, en cas d'utilisation concomitante d'AINS, d'antiagrégants plaquettaires ou d'anticoagulants, et chez les patients ayant été antérieurement traités par radiothérapie pelvienne ou présentant des antécédents digestifs, comme des ulcérations ou des saignements digestifs.
-
Neuropathie périphérique :
- Des cas de neuropathie périphérique, de neuropathie sensitive périphérique (par exemple les paresthésies, dysesthésies) et de neuropathie périphérique motrice ont été observés chez les patients recevant du cabazitaxel. Les patients sous traitement par cabazitaxel doivent informer leur médecin avant de poursuivre le traitement si les symptômes de neuropathie apparaissent, tels qu'une douleur, une brûlure, un picotement, un engourdissement ou une faiblesse. Les médecins doivent évaluer la présence ou l'aggravation de la neuropathie avant chaque traitement. Le traitement doit être retardé jusqu'à amélioration des symptômes. La dose de cabazitaxel doit être réduite de 25 mg/m2 à 20 mg/m2 devant une neuropathie périphérique de grade ≥ 2 persistante (cf Posologie et Mode d'administration).
-
Anémie :
- Des anémies ont été observées chez les patients recevant du cabazitaxel (cf Effets indésirables). Le taux d'hémoglobine et l'hématocrite doivent être contrôlés avant le traitement par cabazitaxel ainsi que lorsque les patients présentent des signes ou symptômes d'anémie ou de perte de sang. Une attention particulière est recommandée chez les patients ayant une hémoglobine < 10 g/dl et des mesures appropriées devront être prises en fonction de la clinique.
-
Risque d'insuffisance rénale :
- Des troubles rénaux associés à des infections, des déshydratations sévères dues à des diarrhées, des vomissements et des uropathies obstructives ont été rapportés.
- Des insuffisances rénales incluant des cas avec une issue fatale ont été observées. Des mesures appropriées doivent être prises pour en identifier la cause et traiter les patients.
- Une hydratation adéquate doit être assurée tout au long du traitement par cabazitaxel. Le patient doit être informé de la nécessité de signaler immédiatement tout changement de diurèse quotidienne. La créatinine plasmatique devra être mesurée à l'initiation, à chaque bilan sanguin, et chaque fois que le patient rapporte une modification de sa diurèse. Le traitement par cabazitaxel doit être interrompu en cas de dégradation de la fonction rénale conduisant à une insuffisance rénale grade ≥ 3 du CTCAE 4.0.
-
Affections respiratoires :
- Des pneumonies interstitielles/pneumopathies inflammatoires et des pneumopathies interstitielles diffuses ont été rapportées et peuvent être associées à une issue fatale (cf Effets indésirables).
- Si de nouveaux symptômes pulmonaires apparaissent ou des symptômes pulmonaires s'aggravent, les patients doivent être surveillés de façon rapprochée, rapidement examinés, et traités de façon appropriée.
- L'interruption de traitement par cabazitaxel est recommandée jusqu'à ce que le diagnostic soit établi. Une prise en charge précoce peut aider à améliorer l'état du patient. Le bénéfice de la reprise du traitement par cabazitaxel doit être évalué avec attention.
-
Risque d'arythmie cardiaque :
- Des arythmies cardiaques ont été rapportées, plus fréquemment des tachycardies et des fibrillations auriculaires (cf Effets indésirables).
-
Patients âgés :
- Les patients âgés (≥ 65 ans) peuvent être plus susceptibles de présenter des effets indésirables incluant des neutropénies et des neutropénies fébriles (cf Effets indésirables).
-
Patients avec insuffisance hépatique :
- Le traitement par Jevtana est contre-indiqué chez les patients ayant une insuffisance hépatique sévère (bilirubine totale > 3 × LSN) (cf Contre-indications, Pharmacocinétique).
- La dose doit être réduite pour les patients ayant une insuffisance hépatique légère (bilirubine totale > 1 à ≤ 1,5 × LSN ou ASAT > 1,5 × LSN) (cf Posologie et Mode d'administration, Pharmacocinétique).
-
Interactions :
- La coadministration d'inhibiteurs puissants du CYP3A doit être évitée car ils peuvent augmenter les concentrations plasmatiques du cabazitaxel (cf Posologie et Mode d'administration, Interactions). Si la coadministration avec un puissant inhibiteur du CYP3A ne peut pas être évitée, une surveillance étroite de la toxicité et une réduction de dose du cabazitaxel devront être envisagées (cf Posologie et Mode d'administration, Interactions).
- La coadministration d'inducteurs puissants du CYP3A doit être évitée car ils peuvent diminuer les concentrations plasmatiques du cabazitaxel (cf Posologie et Mode d'administration, Interactions).
-
Excipients :
- Ce médicament contient 573 mg d'alcool (éthanol) dans chaque flacon de solvant. La quantité contenue dans la dose de ce médicament est équivalente à moins de 11 ml de bière ou 5 ml de vin.
- La faible quantité d'alcool dans ce médicament n'aura pas d'effet notable. Cependant, une précaution particulière est requise chez les groupes à haut risque comme les patients atteints de troubles hépatiques, d'épilepsie et chez les patients ayant des antécédents d'alcoolisme.
-
Contraception :
- Les hommes doivent utiliser une méthode de contraception efficace pendant le traitement et jusqu'à 4 mois après l'arrêt du traitement par cabazitaxel (cf Fertilité/Grossesse/Allaitement).
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Contraception :
En raison du risque génotoxique du cabazitaxel (cf Sécurité préclinique), les hommes doivent utiliser une méthode de contraception efficace pendant le traitement et jusqu'à 4 mois après l'arrêt du traitement par cabazitaxel.
Grossesse :
Il n'y a pas de données sur l'utilisation du cabazitaxel chez la femme enceinte. Des études chez des animaux ont montré une toxicité sur la reproduction à des doses maternotoxiques (cf Sécurité préclinique) et un passage de la barrière placentaire par le cabazitaxel (cf Sécurité préclinique). Comme tous les autres produits cytotoxiques, le cabazitaxel peut nuire au fœtus chez les femmes enceintes exposées. Le cabazitaxel n'est pas indiqué pour une utilisation chez les femmes.
Allaitement :
Des données pharmacocinétiques disponibles chez l'animal ont montré une excrétion du cabazitaxel et de ses métabolites dans le lait maternel (cf Sécurité préclinique).
Fertilité :
Des études chez l'animal ont montré que le cabazitaxel affectait le système de reproduction chez les rats mâles et les chiens sans aucun effet fonctionnel sur la fertilité (cf Sécurité préclinique). Toutefois, compte tenu de l'activité pharmacologique des taxanes, de leur potentiel génotoxique par un mécanisme aneugène et de l'effet de plusieurs composés de cette classe sur la fertilité dans les études animales, l'effet sur la fertilité masculine ne peut être exclu chez l'homme.
Les hommes traités par cabazitaxel sont invités à demander des conseils sur la conservation du sperme avant le traitement.
CONDUITE et UTILISATION DE MACHINES |
Le cabazitaxel a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines car il peut provoquer de la fatigue et des vertiges. Les patients doivent être avisés de ne pas conduire ni d'utiliser des machines s'ils présentent ces effets indésirables pendant le traitement.
Il n'y a pas d'antidote au cabazitaxel. Les complications prévisibles liées au surdosage consisteraient en une exacerbation des effets indésirables, tels qu'une aplasie médullaire et des troubles gastro-intestinaux.
En cas de surdosage, le patient doit être admis dans une unité spécialisée afin de surveiller étroitement ses fonctions vitales. Les patients doivent recevoir un traitement par G-CSF dès que possible après découverte du surdosage. D'autres mesures symptomatiques appropriées doivent être prises.
Des réactions indésirables observées dans les études cliniques mais vues chez le chien après l'administration d'une dose unique, durant 5 jours consécutifs, et un rythme hebdomadaire à des niveaux d'exposition plus faible que les niveaux d'exposition clinique et pouvant être pertinents en clinique, étaient des nécroses artériolaires/périartériolaires dans le foie, des hyperplasies de la voie biliaire et/ou des nécroses hépatocellulaires (cf Posologie et Mode d'administration).
Des réactions indésirables non observées dans les études cliniques, mais vues chez le rat lors d'études de toxicité à doses répétées, à des niveaux d'exposition plus élevés que les niveaux d'exposition clinique et pouvant être pertinents en clinique, étaient des troubles oculaires caractérisés par un gonflement/dégénérescence de la fibre optique sous-capsulaire. Ces effets étaient partiellement réversibles après 8 semaines.
Aucune étude de carcinogénicité n'a été menée avec le cabazitaxel.
Le cabazitaxel n'a pas induit de mutations dans le test de mutation reverse bactérienne (Ames). Il n'était pas clastogénique dans les tests in vitro dans les lymphocytes humains (pas d'induction d'aberration structurale chromosomique mais il augmentait le nombre de cellules polyploïdes) et a induit une augmentation des micronoyaux dans les tests in vivo chez les rats. Ces résultats de génotoxicité (par un mécanisme aneugène) sont inhérents à l'activité pharmacologique de la molécule (inhibition de la dépolymérisation de la tubuline).
Le cabazitaxel n'a pas d'incidence sur les performances d'accouplement ou la fertilité des rats mâles traités. Cependant, dans les études de toxicité à doses répétées, une dégénérescence de la vésicule séminale et une atrophie du tubule séminifère dans les testicules ont été observées chez le rat et une dégénérescence testiculaire (nécrose minime des cellules épithéliales uniques dans l'épididyme) a été observée chez les chiens. Les expositions chez les animaux étaient similaires ou plus faibles que celles vues chez les humains recevant des doses cliniquement pertinentes de cabazitaxel. Le cabazitaxel a induit une toxicité embryofœtale chez le rat femelle traité par voie intraveineuse une fois par jour depuis le 6e jour gestationnel au 17e liée à une toxicité maternelle et consistant à des morts fœtales et une diminution du poids moyen fœtal associée à un retard dans l'ossification du squelette. Les expositions chez l'animal étaient plus basses que celles vues chez les humains recevant des doses cliniquement pertinentes de cabazitaxel. Le cabazitaxel passe la barrière placentaire chez le rat.
Chez les rats, le cabazitaxel et ses métabolites sont excrétés dans le lait maternel à une quantité pouvant aller jusqu'à 1,5 % de la dose administrée sur 24 heures.
-
Évaluation du risque environnemental :
- Les résultats des études d'évaluation du risque environnemental ont indiqué que l'utilisation de Jevtana n'aura pas de risque significatif sur l'environnement aquatique (cf Modalités de manipulation et d'élimination).
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Modalités de manipulation et d'élimination.
Les poches de perfusion en PVC ou les sets de perfusion en polyuréthane ne doivent pas être utilisés pour la préparation et l'administration de la solution pour perfusion.
MODALITÉS DE CONSERVATION |
-
Avant ouverture :
-
Durée de conservation : 3 ans.
A conserver à une température ne dépassant pas 30 °C.
Ne pas conserver au réfrigérateur.
-
Après ouverture :
- Les flacons de solution et de solvant doivent être utilisés immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l'utilisateur.
-
Après la dilution initiale de la solution avec le solvant :
- La stabilité physicochimique a été démontrée pendant 1 heure à température ambiante (15 °C-30 °C). D'un point de vue microbiologique, le mélange solution à diluer et solvant doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l'utilisateur et ne devraient pas normalement dépasser 24 heures à 2 °C-8 °C, sauf en cas de dilution réalisée dans des conditions d'asepsie contrôlées et validées.
-
Après la dilution finale dans la poche de perfusion :
- La stabilité physicochimique de la solution pour perfusion a été démontrée pendant 8 heures à température ambiante (incluant l'heure d'administration de la perfusion) et pendant 48 heures au réfrigérateur (incluant l'heure d'administration de la perfusion). D'un point de vue microbiologique, la solution pour perfusion doit être utilisée immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation relèvent de la responsabilité de l'utilisateur et ne devraient pas normalement dépasser 24 heures à 2 °C-8 °C, sauf en cas de dilution réalisée dans des conditions d'asepsie contrôlées et validées.
MODALITÉS MANIPULATION/ÉLIMINATION |
Jevtana doit être préparé et manipulé seulement par un personnel formé à la manipulation des agents cytotoxiques. Les femmes enceintes ne doivent pas manipuler le médicament. Comme tous les autres agents antinéoplasiques, des précautions doivent être prises pendant la manipulation et la préparation de la solution de Jevtana, prenant en compte l'utilisation des dispositifs adaptés, des équipements de protection personnelle (comme des gants), et des procédures de préparation. En cas de contact cutané lors de chacune des étapes de préparation de Jevtana, laver immédiatement et soigneusement la peau avec de l'eau et du savon. En cas de contact avec une muqueuse, laver immédiatement et soigneusement à grande eau la muqueuse contaminée.
Toujours diluer la solution à diluer avec la totalité du solvant fourni avant de l'ajouter dans la solution de perfusion.
Lisez attentivement la totalité de la rubrique avant d'effectuer les étapes de mélange et de dilution. Jevtana requiert deux dilutions avant administration. Suivez les instructions de préparation mentionnées ci-dessous.
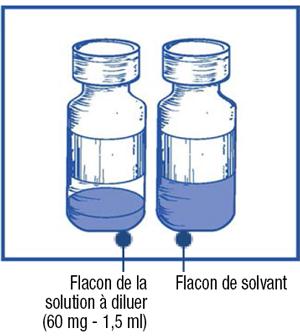
Remarque : le flacon de la solution à diluer de Jevtana 60 mg/1,5 ml (volume de remplissage : 73,2 mg de cabazitaxel/1,83 ml) et le flacon de solvant (volume de remplissage : 5,67 ml) contiennent tous les deux un surremplissage pour compenser la perte de liquide survenant lors de la préparation.
Ce surremplissage permet d'obtenir une solution contenant 10 mg/ml de cabazitaxel après dilution de la solution à diluer de Jevtana avec la totalité du contenu du flacon de solvant fourni.
Les deux étapes suivantes concernant la procédure de dilution de la solution pour perfusion doivent être réalisées de manière aseptique.
-
Étape 1 : Dilution initiale de la solution à diluer avec le solvant fourni :
-
-
Étape 1.1 :
-
- Inspectez visuellement le flacon de la solution à diluer et le flacon de solvant fourni. La solution à diluer et le solvant doivent être limpides.
-
-
Étape 1.2 :
-
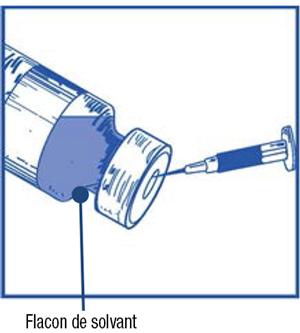
- En utilisant une seringue munie d'une aiguille, prélevez de façon aseptique la totalité du contenu du flacon de solvant fourni en retournant partiellement le flacon.
-
-
Étape 1.3 :
-
- Injectez la totalité du contenu de la seringue dans le flacon de la solution à diluer correspondant.
- Afin de limiter autant que possible la formation de mousse en injectant le solvant, dirigez l'aiguille sur la paroi interne du flacon de la solution à diluer et injectez lentement.
- Une fois reconstituée, la solution obtenue contient 10 mg/ml de cabazitaxel.
-
-
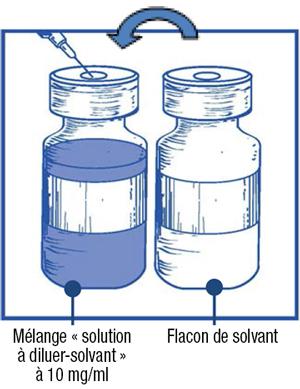
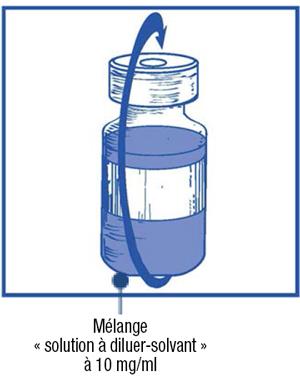
Étape 1.4 :
-
- Retirez la seringue et l'aiguille et mélangez manuellement et doucement par retournements répétés de manière à obtenir une solution claire et homogène. Cela peut prendre environ 45 secondes.
-
-
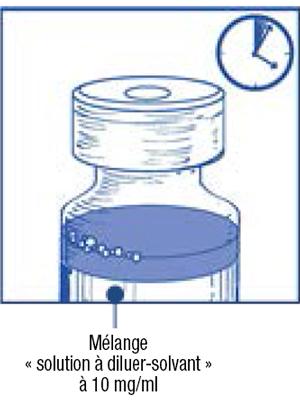
Étape 1.5 :
-
- Laissez reposer cette solution environ 5 minutes puis vérifiez que la solution est homogène et claire. Il est normal que la mousse persiste après cette première étape.
- Ce mélange « solution à diluer-solvant » obtenu contient 10 mg/ml de cabazitaxel (au moins 6 ml de volume extractible). La seconde dilution doit être effectuée immédiatement (dans l'heure) comme détaillé dans l'étape 2.
- Plus d'un flacon de mélange « solution à diluer-solvant » peut être nécessaire pour administrer la dose prescrite.
-
Étape 2 : seconde dilution pour perfusion (dilution finale) :
-
-
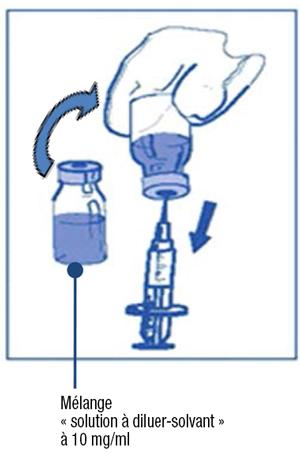
Étape 2.1 :
-
- Prélevez de façon aseptique le volume requis de mélange « solution à diluer-solvant » (contenant 10 mg/ml de cabazitaxel), avec une seringue graduée munie d'une aiguille. A titre d'exemple, une dose de 45 mg de Jevtana nécessiterait 4,5 ml de mélange « solution à diluer-solvant » préparé selon les modalités de l'étape 1.
- Comme la mousse peut persister sur la paroi interne du flacon de cette solution à la suite de la préparation décrite à l'étape 1, il est préférable de placer l'aiguille de la seringue au milieu de la solution lors du prélèvement.
-
-
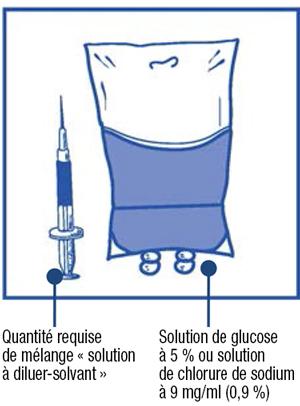
Étape 2.2 :
-
- Injectez le contenu de la seringue dans une poche stérile pour perfusion exempte de PVC contenant soit une solution de glucose à 5 % soit une solution de chlorure de sodium à 9 mg/ml (0,9 %). La concentration de la solution à perfuser doit être comprise entre 0,10 mg/ml et 0,26 mg/ml.
-
-
Étape 2.3 :
-
- Retirez la seringue et mélangez le contenu de la poche ou flacon de perfusion par rotation manuelle.
-
-
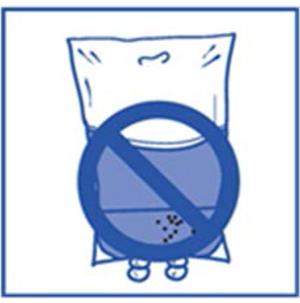
Étape 2.4 :
-
- Comme tous les médicaments administrés par voie parentérale, la solution pour perfusion obtenue doit être contrôlée visuellement avant utilisation. Comme la solution pour perfusion est hypersaturée, elle peut parfois cristalliser avec le temps. Dans ce cas, la solution ne doit pas être utilisée et doit être détruite.
La solution pour perfusion doit être utilisée immédiatement. Toutefois, la durée de conservation peut être plus longue sous certaines conditions précisées dans la rubrique Modalités de conservation.
Un filtre en ligne de pores de 0,22 micromètre de diamètre (communément appelé 0,2 micromètre) est recommandé lors de l'administration.
N'utilisez pas des poches de perfusion en PVC ni de sets de perfusion contenant du polyuréthane pour la préparation et l'administration de Jevtana.
Jevtana ne doit pas être mélangé avec des médicaments autres que ceux mentionnés.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
LISTE I
| Réservé à l'usage hospitalier. |
| Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie. |
| Médicament nécessitant une surveillance particulière pendant le traitement. |
| AMM | EU/1/11/676/001 ; CIP 3400957984977 (RCP rév 30.03.2023). |
| Inscrit sur la liste des spécialités prises en charge en sus des GHS. Collect. |