Classification pharmacothérapeutique VIDAL
Hémostase - Hématopoïèse - Hémoglobinopathies > Stimulants de l'hématopoïèse > Facteurs de croissance plaquettaire (Romiplostim)
Classification ATC
SANG ET ORGANES HEMATOPOIETIQUES > ANTIHEMORRAGIQUES > VITAMINE K ET AUTRES HEMOSTATIQUES > AUTRES HEMOSTATIQUES SYSTEMIQUES (ROMIPLOSTIM)
Excipients
mannitol,
saccharose,
histidine,
acide chlorhydrique,
polysorbate 20 excipient du solvant : eau ppi
Présentation
NPLATE 250 µg Pdre/solv p sol inj Fl+Ser/0,72ml+kit
Cip : 3400935985583
Modalités de conservation : Avant ouverture : < 25° durant 24 heures (A conserver à température ambiante, Conserver à l'abri de la lumière, Conserver dans son emballage), 2° < t < 8° durant 36 mois (Conserver à l'abri de la lumière, Conserver au réfrigérateur, Conserver dans son emballage, Ne pas congeler)
| Poudre : | par flacon |
Romiplostim*
| 250 microgrammes |
| ou | 500 microgrammes |
Excipients (communs) :
mannitol (E421), saccharose, L-histidine, acide chlorhydrique (pour l'ajustement du pH), polysorbate 20.
Solvant : eau pour préparations injectables.
Poudre à 250 microgrammes : Après reconstitution, un volume injectable de 0,5 ml de solution contient 250 microgrammes de romiplostim (500 microgrammes/ml). Chaque flacon contient un surremplissage supplémentaire permettant la délivrance de 250 microgrammes de romiplostim.
Poudre à 500 microgrammes : Après reconstitution, un volume injectable de 1 ml de solution contient 500 microgrammes de romiplostim (500 microgrammes/ml). Chaque flacon contient un surremplissage supplémentaire permettant la délivrance de 500 microgrammes de romiplostim.
*
Le romiplostim est produit par la technique de l'ADN recombinant à partir de cellules d'
Escherichia coli.
Nplate est indiqué chez les patients adultes présentant une thrombopénie immunologique primaire (PTI), réfractaire aux autres traitements (par exemple corticoïdes, immunoglobulines) (
cf Posologie et Mode d'administration, Pharmacodynamie).
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
-
Traçabilité :
- Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
-
Réapparition de la thrombopénie et des saignements après l'interruption du traitement :
- La thrombopénie est susceptible de réapparaître à l'arrêt du traitement par romiplostim. Le risque de saignement augmente si le traitement par romiplostim est interrompu alors qu'un traitement par anticoagulants ou antiagrégants plaquettaires est en cours. Les patients doivent être étroitement surveillés afin de déceler toute diminution du taux de plaquettes et être pris en charge médicalement afin d'éviter tout saignement à l'arrêt du traitement par romiplostim. En cas d'interruption du traitement par romiplostim, il est recommandé de reprendre le traitement du PTI selon les recommandations usuelles de prise en charge. Une prise en charge médicale supplémentaire peut comprendre l'arrêt des anticoagulants et/ou des antiagrégants plaquettaires, des antidotes aux anticoagulants ou des transfusions de plaquettes.
-
Augmentation de la réticuline dans la moelle osseuse :
- L'augmentation de réticuline dans la moelle osseuse semble être le résultat de la stimulation du récepteur à la TPO entraînant une augmentation du nombre de mégacaryocytes dans la moelle osseuse pouvant par la suite induire une libération de cytokines.
- L'augmentation de la réticuline peut être évoquée lors de modifications morphologiques des cellules sanguines périphériques et peut être détectée par une biopsie de la moelle osseuse. Il est donc recommandé de rechercher les anomalies cytologiques sur frottis sanguin et d'effectuer des numérations de la formule sanguine (NFS) avant et pendant le traitement par romiplostim. Cf Effets indésirables pour les informations sur l'augmentation de la réticuline observée dans les essais cliniques avec le romiplostim.
- Si une perte d'efficacité et des anomalies cytologiques sont observées sur le frottis sanguin du patient, l'administration de romiplostim doit être interrompue, un examen clinique doit être effectué et une biopsie de la moelle osseuse avec une coloration appropriée de la réticuline doit être envisagée. La comparaison avec une précédente biopsie de moelle osseuse, si disponible, doit être effectuée. Si l'efficacité est maintenue et si le frottis sanguin observé chez le patient est anormal, le médecin devra réévaluer toutes les options cliniques, notamment la réalisation d'une biopsie de moelle osseuse, la réévaluation du rapport bénéfice/risque de la poursuite du traitement par romiplostim ou l'instauration d'un autre traitement du PTI.
-
Complications thrombotiques/thromboemboliques :
- Un taux de plaquettes supérieur aux valeurs normales expose à un risque de complications thrombotiques/thromboemboliques. L'incidence des évènements thrombotiques/thromboemboliques observés dans les études cliniques était de 6,0 % avec le romiplostim et de 3,6 % avec le placebo. Des précautions sont nécessaires en cas d'administration de romiplostim chez les patients ayant des facteurs de risque connus de thromboembolie incluant, entre autres, des facteurs de risque héréditaires (par exemple Facteur V de Leiden) ou des facteurs de risque acquis (par exemple déficience d'ATIII, syndrome des antiphospholipides), un âge avancé, des patients ayant des périodes prolongées d'immobilisation, des pathologies malignes, des traitements contraceptifs ou hormono-substitutifs, une chirurgie ou un traumatisme, une obésité et un tabagisme.
- Chez des patients atteints de maladie hépatique chronique traités par romiplostim, des cas d'évènements thromboemboliques, incluant des thromboses de la veine porte, ont été rapportés. Le romiplostim doit être utilisé avec précaution chez ces patients. Les recommandations d'ajustement de dose doivent être suivies (cf Posologie et Mode d'administration).
-
Erreurs médicamenteuses :
- Des erreurs médicamenteuses, incluant des surdosages et des sous-dosages, ont été rapportées chez des patients recevant Nplate ; les recommandations concernant le calcul de la dose et l'ajustement posologique doivent être suivies (cf Posologie et Mode d'administration).
- Le surdosage peut entraîner une augmentation excessive des taux de plaquettes associée à des complications thrombotiques/thromboemboliques. Si le taux de plaquettes augmente de façon excessive, arrêter Nplate et surveiller le taux de plaquettes. Reprendre le traitement par Nplate en suivant les recommandations de posologie et d'administration. Le sous-dosage peut se traduire par un taux de plaquettes plus bas que prévu et un risque potentiel de saignement. Le taux de plaquettes doit être surveillé chez les patients recevant Nplate (cf Posologie et Mode d'administration, Mises en garde et Précautions d'emploi, Surdosage).
-
Progression des syndromes myélodysplasiques (SMD) existants :
- Un rapport bénéfice/risque favorable du romiplostim a été établi uniquement pour le traitement de la thrombopénie associée au PTI (cf Mises en garde et Précautions d'emploi) et le romiplostim ne doit pas être utilisé dans d'autres situations cliniques associées à une thrombopénie.
- Chez les adultes et les patients âgés, le diagnostic de PTI doit avoir été confirmé par l'exclusion d'autres situations cliniques pouvant induire une thrombopénie, en particulier le diagnostic d'un SMD doit être éliminé. La réalisation d'une ponction et d'une biopsie de la moelle osseuse doit normalement avoir été réalisée au cours de la maladie et du traitement, en particulier chez les patients âgés de plus de 60 ans, et ceux présentant des symptômes systémiques ou des signes anormaux tels qu'une augmentation des cellules blastiques périphériques.
- Au cours d'études cliniques évaluant le romiplostim chez des patients présentant un SMD, des cas d'augmentation transitoire des cellules blastiques ont été observés et des cas de progression de SMD en leucémie aiguë myéloïde (LAM) ont été signalés. Au cours d'un essai clinique randomisé, contrôlé versus placebo, chez des patients présentant un SMD, le traitement par le romiplostim a été prématurément arrêté en raison d'un excès du nombre de cas de progression de SMD en LAM et d'une augmentation du nombre de cellules blastiques circulantes de plus de 10 % chez les patients recevant du romiplostim par rapport au placebo. Parmi les cas de progression de SMD en LAM qui ont été observés, les patients dont le SMD était initialement classé RAEB-1 étaient plus sujets à une progression en LAM par rapport aux SMD de plus faible risque.
- Le romiplostim ne doit pas être utilisé pour le traitement des thrombopénies dues aux SMD ou toute autre cause de thrombopénie autre que le PTI en dehors des essais cliniques.
-
Perte de réponse au romiplostim :
- Une diminution de la réponse ou un échec de maintien d'une réponse plaquettaire avec un traitement par romiplostim dans l'intervalle des posologies recommandées doit inciter à une recherche des facteurs étiologiques, incluant l'immunogénicité (cf Effets indésirables) et l'augmentation de réticuline dans la moelle osseuse (cf ci-dessus).
-
Effets du romiplostim sur les globules rouges et les globules blancs :
- Des modifications des taux de globules rouges (diminution) et de globules blancs (augmentation) ont été observées dans les études précliniques de toxicologie (chez le rat et le singe) ainsi que chez les patients présentant un PTI. Une anémie et une leucocytose concomitantes peuvent survenir (dans une fenêtre de 4 semaines) chez des patients, indépendamment du fait qu'ils soient splénectomisés ou non, mais ont été observées plus fréquemment chez les patients ayant subi une splénectomie au préalable. La surveillance de ces taux doit être envisagée chez les patients traités par romiplostim.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Grossesse :
Il n'existe pas de données ou des données limitées sur l'utilisation du romiplostim chez la femme enceinte.
Les études effectuées chez l'animal ont montré que le romiplostim passe la barrière placentaire et augmente le taux de plaquettes chez le fœtus. Des pertes post-implantatoires et une légère augmentation de la mortalité périnatale des nouveau-nés ont également été constatées dans les études menées chez l'animal (cf Sécurité préclinique).
Le romiplostim n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement :
On ne sait pas si le romiplostim ou ses métabolites sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. La décision d'interrompre l'allaitement ou d'interrompre/de s'abstenir du traitement par romiplostim doit être prise en prenant en compte le bénéfice de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère.
Fertilité :
Il n'existe pas de données sur la fertilité.
CONDUITE et UTILISATION DE MACHINES |
Nplate a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Dans les essais cliniques, des épisodes transitoires de sensations vertigineuses, de légères à modérées, ont été ressentis par certains patients.
Aucun effet indésirable n'a été observé chez le rat après une dose unique de 1000 microgrammes/kg ou chez le singe après des doses répétées de romiplostim de 500 microgrammes/kg (respectivement 100 ou 50 fois la dose clinique maximale de 10 microgrammes/kg).
En cas de surdosage, le taux de plaquettes peut augmenter de façon excessive et entraîner des complications thrombotiques/thromboemboliques. Si le taux de plaquettes augmente de façon excessive, arrêter Nplate et surveiller le taux de plaquettes. Reprendre le traitement par Nplate en suivant les recommandations de posologie et d'administration (cf Posologie et Mode d'administration, Mises en garde et Précautions d'emploi).
La toxicité à doses répétées du romiplostim a été étudiée sur 4 semaines chez le rat et jusqu'à 6 mois chez le singe. Les effets observés au cours de ces études étaient liés à l'activité thrombopoïétique du romiplostim et étaient comparables, quelle que soit la durée de l'étude. Des réactions au site d'injection ont été observées, liées à l'administration du romiplostim. Des myélofibroses ont été observées dans la moelle osseuse de rats à toutes les doses testées. Au cours de ces études, les myélofibroses ont régressé chez les animaux en 4 semaines, indiquant une réversibilité de cet effet.
Dans une étude toxicologique d'un mois chez le rat et le singe, une diminution modérée du taux de globules rouges, de l'hématocrite et de l'hémoglobine a été observée. Une stimulation de la production de leucocytes a également été constatée comme les taux sériques de neutrophiles, lymphocytes, monocytes et éosinophiles ont modérément augmenté. Dans l'étude la plus longue menée chez le singe, à doses répétées, il n'y a pas eu d'effet sur les lignées érythrocytaire et leucocytaire lors de l'administration du romiplostim pendant 6 mois au cours desquels le schéma d'administration est passé de trois fois par semaine à une fois par semaine. De plus, dans les études pivots de phase III, le romiplostim n'a pas eu d'effet sur les lignées érythrocytaire et leucocytaire par rapport aux patients ayant reçu le placebo.
Du fait de la formation d'anticorps neutralisants chez le rat, une diminution des effets pharmacodynamiques du romiplostim était souvent observée après administration prolongée. Les études de toxicocinétique n'ont pas montré de relation entre les anticorps et les concentrations mesurées. Bien que de fortes doses aient été testées chez l'animal, les marges de sécurité ne peuvent être estimées de façon fiable chez l'homme du fait de différences de sensibilité vis-à-vis de la pharmacodynamie du romiplostim et des effets des anticorps neutralisants.
-
Cancérogénicité :
- Aucune étude spécifique n'a été menée pour étudier le potentiel cancérogène du romiplostim. Le risque cancérogène du romiplostim chez l'homme demeure donc inconnu.
-
Toxicité sur la reproduction :
- Au cours de toutes les études portant sur le développement, la formation d'anticorps neutralisants a été observée ; ceux-ci ont pu inhiber les effets du romiplostim. Les études de développement embryo-fœtal chez la souris et le rat montrent une réduction du poids maternel chez la souris uniquement. Chez la souris, une augmentation des avortements précoces a été mise en évidence. Une augmentation de la durée de la gestation et une légère augmentation de l'incidence de la mortalité périnatale ont été observées au cours d'une étude de développement pré et postnatal chez le rat. Le romiplostim passe la barrière placentaire chez le rat, et pourrait être transmis de la mère au fœtus et stimuler la production de plaquettes chez le fœtus. Le romiplostim n'a montré aucun effet sur la fertilité chez le rat.
Ce médicament ne doit pas être mélangé avec d'autres médicaments, à l'exception de ceux mentionnés dans la rubrique Modalités de manipulation et d'élimination.
MODALITÉS DE CONSERVATION |
Durée de conservation : 3 ans.
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. A conserver dans l'emballage extérieur d'origine, à l'abri de la lumière.
Peut être retiré du réfrigérateur pour une période de 30 jours à température ambiante (jusqu'à 25 °C) lorsque conservé dans l'emballage d'origine.
Après reconstitution :
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 24 heures à 25 °C et pendant 24 heures entre 2 °C et 8 °C, à l'abri de la lumière et conservé dans le flacon d'origine.
D'un point de vue microbiologique, le médicament doit être utilisé immédiatement après reconstitution. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur et ne doivent normalement pas excéder 24 heures à 25 °C ou 24 heures au réfrigérateur (entre 2 °C et 8 °C), à l'abri de la lumière.
MODALITÉS MANIPULATION/ÉLIMINATION |
Nplate est un médicament stérile sans conservateur destiné seulement à un usage unique. Nplate doit être reconstitué dans des conditions d'asepsie rigoureuses.
-
Nplate 250 microgrammes poudre et solvant pour solution injectable :
Nplate 250 microgrammes poudre pour solution injectable doit être reconstitué avec 0,72 ml d'eau stérile pour préparations injectables, donnant un volume injectable de 0,5 ml. Chaque flacon contient un surremplissage supplémentaire permettant d'assurer l'administration de la dose de 250 microgrammes de romiplostim (voir tableau « Contenu du flacon » ci-dessous).
-
Nplate 500 microgrammes poudre et solvant pour solution injectable :
Nplate 500 microgrammes poudre pour solution injectable doit être reconstitué avec 1,2 ml d'eau stérile pour préparations injectables, donnant un volume injectable de 1 ml. Chaque flacon contient un surremplissage supplémentaire permettant d'assurer l'administration de la dose de 500 microgrammes de romiplostim (voir tableau « Contenu du flacon » ci-dessous).
Contenu du flacon :
| Nplate flacon à usage unique | Teneur totale en romiplostim par flacon | | Volume d'eau stérile pour préparations injectables | | Produit délivré et volume | Concentration finale |
| 250 microgrammes | 375 microgrammes | + | 0,72 ml | = | 250 microgrammes dans 0,50 ml | 500 microgrammes/ml |
| 500 microgrammes | 625 microgrammes | + | 1,2 ml | = | 500 microgrammes dans 1,00 ml | 500 microgrammes/ml |
D'un point de vue microbiologique, le produit doit être utilisé immédiatement après reconstitution. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur et ne doivent normalement pas excéder 24 heures à 25 °C ou 24 heures au réfrigérateur (entre 2 °C et 8 °C), à l'abri de la lumière.
- Retirer le capuchon en plastique du flacon de poudre de Nplate et nettoyer le bouchon en caoutchouc à l'aide des compresses alcoolisées fournies.
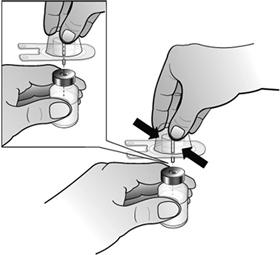
- Fixer l'adaptateur de flacon au flacon de Nplate en retirant la pellicule protectrice de l'adaptateur de flacon, tout en veillant à le maintenir dans son emballage. Tout en maintenant le flacon posé à plat, enfoncer fermement l'adaptateur au centre du flacon jusqu'à ce qu'il soit bien en place.
Remarque : Afin d'éviter toute contamination du médicament, ne pas toucher la pointe de l'adaptateur ou Luer-lock.
-
Retirer et jeter l'emballage de l'adaptateur de flacon.
-
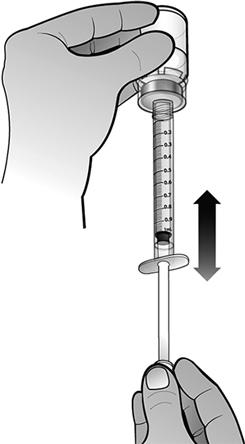
Fixer le piston à la seringue préremplie d'eau pour préparations injectables en tournant le piston dans le sens des aiguilles d'une montre jusqu'à sentir une légère résistance.
-
En tenant la seringue préremplie d'eau pour préparations injectables dans une main, replier l'extrémité de la protection en plastique blanc, avec l'autre main. Ceci brisera le scellé de la protection en plastique blanc. Une fois ce scellé brisé, retirer la protection pour séparer le capuchon en plastique gris de l'extrémité en plastique clair de la seringue.
-
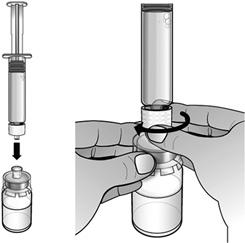
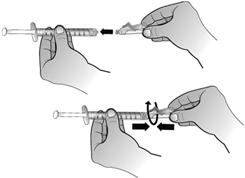
En maintenant le flacon posé à plat, fixer la seringue préremplie d'eau pour préparations injectables à l'adaptateur de flacon : Tenir le bord extérieur de l'adaptateur de flacon d'une main et tourner l'embout de la seringue dans le sens des aiguilles d'une montre dans l'adaptateur avec l'autre main, jusqu'à sentir une légère résistance.
-
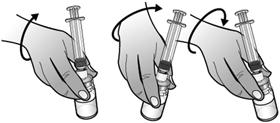
Expulser lentement et délicatement l'eau dans le flacon de poudre. L'eau doit couler lentement sur la poudre. Remuer DELICATEMENT le flacon jusqu'à dissolution complète de la poudre et jusqu'à ce que le liquide soit limpide et incolore.
Ne pas secouer le flacon.
Remarque : D'un point de vue microbiologique, le médicament doit être utilisé immédiatement après reconstitution. S'il n'est pas utilisé immédiatement, la seringue ne doit pas être retirée de l'adaptateur de flacon afin de maintenir une intégrité microbiologique.
Remarque : La dissolution complète de la poudre peut prendre jusqu'à 2 minutes.
Avant de poursuivre :
Inspecter visuellement la solution reconstituée pour rechercher des particules et/ou une coloration. La solution reconstituée doit être limpide et incolore et ne doit pas être administrée si l'on observe des particules et/ou un changement de coloration.
S'assurer que la poudre est totalement dissoute avant de retirer la seringue.
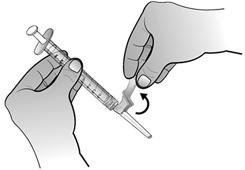
- Retirer la seringue préremplie vide de l'adaptateur de flacon.
-
Retirer la seringue de 1 ml de son emballage. Fixer cette seringue de 1 ml à l'adaptateur de flacon de la solution reconstituée en tournant l'embout de la seringue dans l'adaptateur de flacon jusqu'à sentir une légère résistance.
-
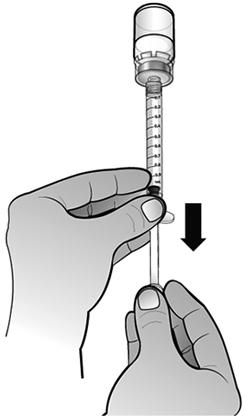
Retourner l'ensemble seringue-flacon afin que le flacon de la solution reconstituée se retrouve au-dessus de la seringue. Prélever tout le volume de la solution reconstituée dans la seringue destinée à l'administration.
Assurez-vous que le piston reste dans la seringue.
-
S'assurer que le volume de solution dans la seringue d'administration correspond à la dose prescrite pour le patient, en injectant l'excès de solution dans le flacon.
Remarque : Éliminer toutes les bulles d'air de la seringue afin d'avoir le volume exact de solution dans la seringue.
-
Retirer la seringue destinée à l'administration de l'adaptateur de flacon.
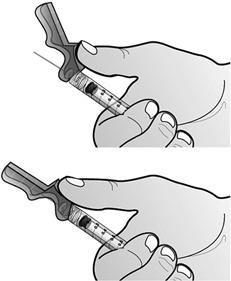
Fixer l'aiguille sécurisée à la seringue destinée à l'administration préalablement remplie en tournant l'aiguille dans le sens des aiguilles d'une montre dans l'embout Luer-lock.
- Préparer le site d'injection à l'aide d'une compresse alcoolisée. Relever le système de sécurité rose vers la seringue en dégageant l'aiguille.
Retirer le capuchon de l'aiguille en tenant la seringue d'une main et en tirant soigneusement sur le capuchon, sans le tourner, de l'autre main.
-
Injecter par voie sous-cutanée conformément aux bonnes pratiques en vigueur.
-
Après injection, activer le système de sécurité rose en poussant le cran de sécurité avec la même main jusqu'à entendre un clic et/ou jusqu'à ressentir la fermeture.
-
Éliminer immédiatement la seringue et l'aiguille dans des conteneurs pour matériel médical usagé prévus à cet effet.
Pour les conditions de conservation après reconstitution du produit, cf Modalités de conservation.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
LISTE I
| Médicament soumis à prescription hospitalière. |
| Prescription réservée aux spécialistes en hématologie, en médecine interne, ou aux médecins compétents en maladie du sang. |
| Médicament nécessitant une surveillance particulière pendant le traitement. |
| AMM | EU/1/08/497/005 ; CIP 3400935985583 (RCP rév 22.01.2021) 250 microgrammes poudre et solvant. |
| EU/1/08/497/007 ; CIP 3400935996411 (RCP rév 22.01.2021) 500 microgrammes poudre et solvant. |
| | |
| Prix : | 584,03 euros (1 flacon à 250 microgrammes + solvant). |
| 1133,05 euros (1 flacon à 500 microgrammes + solvant). |
| Remb Séc soc à 65 %. Collect. |
|
Non remboursable et non agréé Collect, à la date du 19.04.2022, pour les patients adultes présentant un PTI d'une durée d'évolution > 12 mois non splénectomisés ne faisant pas l'objet d'une contre-indication à la splénectomie, et pour les patients adultes présentant un PTI d'une durée d'évolution ≤ 12 mois.
|
Titulaire de l'AMM : Amgen Europe B.V., Minervum 7061, 4817 ZK Breda, Pays-Bas.