Mise à jour : 22 août 2024
Sommaire
SYNTHÈSE |
excipient et excipient du solvant : mannitol
excipient du solvant : sodium hydroxyde, acide chlorhydrique, eau ppi
EEN sans dose seuil : caoutchouc
Cip : 3400930166246
Modalités de conservation : Avant ouverture : < 25° durant 36 mois (Ne pas congeler)
FORMES et PRÉSENTATIONS |
Poudre (poudre blanche à blanchâtre) et solvant (solution limpide, incolore, exempt de particules visibles [pH 5,0-7,0]) pour suspension injectable à libération prolongée.
Kit d'injection contenant :
COMPOSITION |
Chaque flacon contient 3,75 mg d'acétate de leuproréline (équivalent à 3,57 mg de leuproréline sous forme de base libre).
1 mL de suspension reconstituée contient 1,875 mg d'acétate de leuproréline.
Excipients du lyophilisat (flacon) : polysorbate 80, mannitol (E421), carmellose sodique (E466), citrate de triéthyle, polymère d'acide D,L-lactique et d'acide glycolique (PLGA).
Excipients du solvant (seringue préremplie) : mannitol (E421), hydroxyde de sodium (pour ajustement du pH), acide chlorhydrique (pour ajustement du pH), eau pour préparations injectables.
INDICATIONS |
Cancer de la prostate
ZEULIDE 3,75 mg est indiqué dans le traitement palliatif du cancer de la prostate hormono-dépendant à un stade avancé.
Fibromes utérins
ZEULIDE 3,75 mg est indiqué dans le traitement du léiomyome utérin (fibrome utérin). Ce traitement peut être utilisé avant une chirurgie ou en complément d'une chirurgie, ou en tant qu'alternative définitive aux symptômes chez les femmes en péri-ménopause qui ne souhaitent pas d'intervention chirurgicale.
Endométriose
ZEULIDE 3,75 mg est indiqué dans le traitement de l'endométriose. Il peut être utilisé en tant que traitement unique ou en tant que complément à une chirurgie.
Cancer du sein
ZEULIDE 3,75 mg est indiqué dans le traitement des femmes pré- et péri-ménopausées souffrant d'un cancer du sein à un stade avancé et se prêtant à une manipulation hormonale.
ZEULIDE 3,75 mg est indiqué en tant que traitement adjuvant, en association à du tamoxifène ou à un inhibiteur de l'aromatase, du cancer du sein à un stade précoce répondant à l'hormonothérapie chez les femmes pré- et péri-ménopausées à haut risque de récidive (âge jeune, tumeur de haut grade, envahissement ganglionnaire). Chez les femmes ayant reçu une chimiothérapie, le statut pré-ménopausique doit être confirmé avant la fin de la chimiothérapie.
Protection ovarienne
ZEULIDE 3,75 mg est indiqué pour la préservation de la fonction ovarienne chez les femmes pré-ménopausées présentant une maladie néoplasique et recevant une chimiothérapie susceptible de provoquer une insuffisance ovarienne prématurée.
Puberté précoce centrale
ZEULIDE 3,75 mg est également indiqué dans le traitement des enfants présentant un diagnostic de puberté précoce centrale (PPC) confirmé par l'apparition de caractéristiques sexuelles secondaires avant l'âge de neuf ans chez les fillettes et dix ans chez les garçons.
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
CONTRE-INDICATIONS |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Généralités
Des convulsions ont été rapportées suite à l'administration d'acétate de leuproréline. Ces cas ont été observés chez des patients ayant des antécédents de crises de convulsions, d'épilepsie, de troubles cérébrovasculaires, d'anomalies ou de tumeurs du système nerveux central, et chez les patients prenant des traitements concomitants ayant été associés aux crises tels que le bupropion et les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS). Des convulsions ont également été rapportées chez des patients en cas d'absence d'une quelconque maladie mentionnée ci-dessus.
Il existe un risque accru d'épisode de dépression (pouvant être sévère) chez les patients sous traitement par agonistes de la GnRH, tels que l'acétate de leuproréline. Les patients doivent être informés en conséquence et convenablement traités si des symptômes apparaissent.
Le traitement doit être immédiatement interrompu si le patient présente un quelconque signe ou symptôme indiquant une anaphylaxie/une réaction anaphylactique (dyspnée, asthme, rhinite, œdème angioneurotique ou de la glotte, hypotension, urticaire, éruption cutanée, prurit ou pneumonie interstitielle). Les patients doivent être informés avant l'instauration du traitement, qu'en cas d'apparition de l'un des symptômes mentionnés précédemment, ils doivent arrêter le traitement et consulter leur médecin. Les patients ayant présenté une réaction d'hypersensibilité à la leuproréline doivent être étroitement surveillés et ne doivent pas reprendre ZEULIDE 3,75 mg.
Des cas de dysfonctionnement hépatique et de jaunisse accompagnés d'un taux élevé d'enzymes hépatiques ont été rapportés avec l'utilisation de l'acétate de leuproréline. Par conséquent, une surveillance étroite et les mesures appropriées doivent être mises en place si nécessaire.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c'est-à-dire qu'il est essentiellement « sans sodium ».
Hypertension intracrânienne idiopathique
Des cas d'hypertension intracrânienne idiopathique (méningite séreuse) ont été rapportés chez des patients recevant de la leuproréline. Les patients doivent être avertis de la possibilité de signes et symptômes d'hypertension intracrânienne idiopathique, notamment des céphalées sévères ou récurrentes, de troubles visuels et d'acouphènes. En présence d'une hypertension intracrânienne idiopathique, l'interruption du traitement par leuproréline doit être envisagée.
Concernant les hommes
Lors des premières phases du traitement par ZEULIDE 3,75 mg, comme lors des traitements avec d'autres agonistes de la GnRH, une augmentation transitoire des taux de testostérone peut se produire. Dans certains cas, ceci peut être associé à une « poussée » ou à une exacerbation de la croissance tumorale qui se traduit par une aggravation temporaire des symptômes du cancer de la prostate. Ces symptômes se dissipent généralement en poursuivant le traitement (voir rubrique Effets indésirables). La « poussée » peut parfois se manifester par des symptômes systémiques ou neurologiques (par ex. douleurs osseuses, etc.). De plus, des cas d'atrophie testiculaire et de gynécomastie ont été également décrits avec d'autres agonistes de la GnRH.
Chez les patients traités avec de l'acétate de leuproréline, des cas isolés d'obstruction urétérale (avec ou sans hématurie) et de compression de la moelle épinière ou de lésions métastatiques vertébrales ont été observés, pouvant entraîner une paralysie avec ou sans complications fatales. Les patients risquant de présenter une obstruction urétérale, une compression de la moelle épinière ou des lésions métastatiques vertébrales doivent être soigneusement examinés et étroitement surveillés durant les premières semaines du traitement. Un traitement prophylactique avec des antiandrogènes doit être envisagé pour ces patients.
Des complications urologiques ou neurologiques peuvent survenir, celles-ci doivent être traitées par des mesures spécifiques appropriées.
Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez les hommes ayant subi une orchidectomie ou ayant été traités par un agoniste de la GnRH. L'ajout d'un antiandrogène au traitement peut réduire la perte osseuse mais augmente le risque d'autres effets indésirables tels que des problèmes de coagulation et des œdèmes. Lorsqu'un antiandrogène est utilisé sur une longue période, il convient d'apporter une attention particulière aux contre-indications et aux précautions associées à son utilisation prolongée. Les patients à risque ou avec des antécédents d'ostéoporose doivent être soigneusement examinés et étroitement surveillés pendant le traitement par acétate de leuproréline (voir rubrique Effets indésirables).
La réponse au traitement par ZEULIDE 3,75 mg doit être surveillée en procédant à des examens cliniques et à l'analyse régulière des taux sériques de testostérone et du PSA.
Certains patients peuvent présenter des changements métaboliques (par ex. intolérance au glucose ou aggravation d'un diabète existant), une hypertension, des changements de poids et des troubles cardiovasculaires. Comme attendu avec ce type de médicament, un développement ou une aggravation du diabète peut survenir et par conséquent, les patients diabétiques peuvent nécessiter une surveillance de la glycémie plus fréquente pendant le traitement par ZEULIDE 3,75 mg. Les patients à haut risque de présenter des maladies métaboliques ou cardiovasculaires doivent être soigneusement évalués avant de commencer le traitement et correctement surveillés pendant le traitement par privation androgénique. Le traitement par acétate de leuproréline entraîne une suppression du système hypophyso-gonadique. Les résultats des tests diagnostiques des fonctions de la gonadostimuline hypophysaire et des gonades effectués pendant et après le traitement par acétate de leuproréline peuvent être altérés.
Une augmentation du temps de prothrombine a été rapportée chez les patients traités avec de l'acétate de leuproréline.
L'acétate de leuproréline doit être utilisé avec précaution en cas de maladie cardiovasculaire (y compris l'insuffisance cardiaque congestive), de thromboembolie, d'œdème, de dépression et d'apoplexie hypophysaire.
L'acétate de leuproréline doit être utilisé avec précaution chez les patients présentant des troubles sanguins, une thrombocytopénie ou prenant un traitement anticoagulant.
Une thérapie par privation androgénique peut allonger l'intervalle QT
Chez les patients ayant des antécédents ou des facteurs de risque d'allongement de l'intervalle QT, de même que chez les patients traités en concomitance avec des substances connues pour allonger l'intervalle QT (voir rubrique Interactions), les médecins doivent évaluer le rapport bénéfices-risques, y compris la possibilité de torsades de pointe, avant d'instaurer un traitement par ZEULIDE 3,75 mg.
Concernant les femmes
Étant donné que les règles doivent normalement cesser lorsque l'on utilise des doses efficaces de ZEULIDE 3,75 mg, la patiente doit avertir son médecin si des règles régulières persistent.
Étant donné qu'une perte de la densité osseuse est attendue dans le cadre d'une ménopause naturelle, celle-ci devrait également être observée lors de l'induction médicale d'un état hypo-œstrogénique. Cette perte osseuse s'est révélée réversible après l'arrêt d'un traitement de six mois par acétate de leuproréline.
Pendant la première phase du traitement, les stéroïdes sexuels augmentent temporairement au-dessus des valeurs initiales en raison de l'effet physiologique du médicament. Il est dès lors possible d'observer une augmentation des signes et symptômes cliniques pendant les premiers jours de traitement, mais ceux-ci disparaissent lorsque le traitement est poursuivi aux doses adéquates. On a toutefois rapporté des saignements vaginaux abondants nécessitant une intervention médicale ou chirurgicale lors de la poursuite du traitement lorsque le médicament était utilisé pour le traitement de léiomyomes utérins sous-muqueux.
L'utilisation sûre de l'acétate de leuproréline pendant la grossesse n'a pas été établie cliniquement. Avant d'instaurer un traitement par acétate de leuproréline, il est conseillé de déterminer si la patiente est enceinte. La leuproréline n'est pas un contraceptif. Si une contraception est nécessaire, une méthode non hormonale doit être utilisée.
Cancer du sein
Afin de garantir une suppression ovarienne adéquate chez les femmes pré- et péri-ménopausées, le traitement par leuproréline doit être administré pendant au moins 6 à 8 semaines avant de débuter l'inhibiteur de l'aromatase, et les injections mensuelles de leuproréline doivent être administrées conformément au calendrier prévu et sans interruption pendant toute la durée du traitement par l'inhibiteur de l'aromatase.
Les femmes pré-ménopausées au moment du diagnostic du cancer du sein et qui deviennent aménorrhéiques après la chimiothérapie peuvent continuer ou non à produire des œstrogènes. Quel que soit leur statut menstruel, le statut pré-ménopausique doit être confirmé, après la chimiothérapie et avant de débuter la leuproréline, en s'assurant que les concentrations sanguines d'œstradiol et de FSH se situent dans les valeurs de référence pour les femmes pré-ménopausées, afin d'éviter tout traitement inutile par leuproréline chez une femme qui présenterait une ménopause chimio-induite.
Après avoir commencé la leuproréline, il est important de confirmer qu'il y a bien suppression ovarienne (ménopause induite par un analogue de la gonadotrophine) en réalisant des dosages sériés des taux circulants de FSH et d'œstradiol si ce sous-groupe de femmes entre en considération pour le traitement par un inhibiteur de l'aromatase selon les recommandations cliniques actuelles. Par conséquent, la suppression ovarienne doit être confirmée par la détermination de concentrations sanguines faibles de FSH et d'œstradiol avant le début du traitement par l'inhibiteur de l'aromatase et ces analyses doivent être répétées tous les trois mois pendant toute la durée du traitement combiné par la leuproréline et un inhibiteur de l'aromatase. Ceci, pour éviter toute augmentation rebond des taux circulants d'œstrogènes, induite par l'inhibiteur de l'aromatase et pouvant avoir des conséquences éventuelles pour le cancer du sein. Il est à noter que les taux circulants de FSH sont réduits en réponse à la suppression ovarienne induite par l'analogue de la gonadotrophine (ménopause induite), contrairement à ce qui se passe au cours de la ménopause naturelle, où les taux de FSH sont élevés.
Les patientes qui ont arrêté le traitement par leuproréline doivent également arrêter les inhibiteurs de l'aromatase dans le mois qui suit la dernière administration de leuproréline. Une attention particulière doit également être accordée aux informations de prescription des médicaments administrés conjointement, tels que les inhibiteurs de l'aromatase, le tamoxifène, les inhibiteurs de CDK4/6, pour toute information pertinente relative à la sécurité lorsque ces produits sont administrés avec la leuproréline.
Il convient d'évaluer la densité minérale osseuse avant d'instaurer le traitement par leuproréline, en particulier chez les femmes qui présentent d'autres facteurs de risque de l'ostéoporose. Aucune donnée spécifique n'est disponible concernant les patientes ayant une ostéoporose établie ou des facteurs de risque d'ostéoporose (p. ex., consommation abusive chronique d'alcool, fumeuses, traitement à long terme de médicaments qui réduisent la densité minérale osseuse, comme les anticonvulsivants ou les corticoïdes, antécédents familiaux d'ostéoporose, malnutrition, comme l'anorexie). Puisqu'une diminution de la densité minérale osseuse est susceptible d'être plus préjudiciable chez ces patientes, le traitement par leuproréline doit être envisagé au cas par cas et ne doit être instauré qu'après avoir soigneusement évalué les bénéfices du traitement par rapport au risque. Ces patientes doivent être surveillées étroitement et un traitement ou une prophylaxie de l'ostéoporose doit être instauré(e) si nécessaire.
Lorsqu'un agoniste de la GnRH est utilisé conjointement à un inhibiteur de l'aromatase ou à du tamoxifène, le risque de troubles musculo-squelettiques (notamment des douleurs articulaires ou musculo-squelettiques) est d'environ 89 % avec l'inhibiteur de l'aromatase et 76 % avec le tamoxifène.
Une hypertension a été rapportée comme étant un effet indésirable ciblé, à une fréquence définie comme « très fréquente », en cas d'association d'un agoniste de la GnRH avec de l'exémestane ou du tamoxifène.
Les femmes pré-ménopausées atteintes d'un cancer du sein et recevant un agoniste de la GnRH en association avec de l'exémestane ou du tamoxifène doivent faire l'objet d'une surveillance régulière des facteurs de risque cardiovasculaires et de la pression sanguine.
Une hyperglycémie et un diabète ont été signalés en tant qu'effets indésirables ciblés, à une fréquence définie comme « fréquente », en cas d'association d'un agoniste de la GnRH avec de l'exémestane ou du tamoxifène. Les femmes pré-ménopausées atteintes d'un cancer du sein et recevant un agoniste de la GnRH en association avec de l'exémestane ou du tamoxifène doivent faire l'objet d'une surveillance régulière des facteurs de risque du diabète par le biais d'une surveillance régulière de la glycémie et l'instauration d'un traitement antidiabétique approprié, si nécessaire, conformément aux recommandations nationales.
Une dépression a été rapportée chez environ 50 % des patientes traitées par un agoniste de la GnRH en association avec du tamoxifène ou de l'exémestane, mais moins de 5 % des patientes ont présenté une dépression sévère (grade 3-4). Les patientes doivent être informées en conséquence et traitées si des symptômes surviennent. Les patientes ayant une dépression connue ou des antécédents de dépression doivent faire l'objet d'une surveillance attentive pendant le traitement.
Le traitement des femmes pré-ménopausées atteintes d'un cancer du sein à un stade précoce répondant à l'hormonothérapie et traitées par leuproréline en association avec du tamoxifène ou un inhibiteur de l'aromatase doit se faire sur base d'une évaluation minutieuse des risques et des bénéfices.
Population pédiatrique
Avant d'instaurer le traitement, un diagnostic précis de la puberté précoce centrale idiopathique et/ou neurogène est nécessaire.
Il s'agit d'un traitement à long terme, qui est ajusté individuellement. ZEULIDE 3,75 mg doit être administré selon un calendrier le plus précis possible à intervalles mensuels réguliers. Le fait de retarder exceptionnellement la date d'injection de quelques jours (30 ± 2 jours) n'influence pas le résultat du traitement.
Dans le cas d'un abcès stérile au site d'injection (rapporté le plus fréquemment après l'injection IM d'une dose plus élevée que celle recommandée), il est possible que l'absorption d'acétate de leuproréline à libération prolongée soit réduite. Dans ce cas, les paramètres hormonaux (testostérone, œstradiol) doivent être contrôlés toutes les deux semaines (voir rubrique Posologie et mode d'administration).
Le traitement des enfants qui présentent des tumeurs cérébrales évolutives doit se faire sur base d'une évaluation minutieuse des risques et des bénéfices.
Chez les fillettes, la survenue de saignements vaginaux, d'un spotting et de pertes vaginales après la première injection peut être le signe d'un retrait hormonal. Des saignements vaginaux survenant au-delà du premier/deuxième mois de traitement doivent être investigués.
La densité minérale osseuse (DMO) peut diminuer pendant un traitement de la puberté précoce par une GnRHa. Toutefois, à l'arrêt du traitement, la masse osseuse accumulée par la suite est maintenue et le pic de masse osseuse atteint à la fin de l'adolescence ne semble pas affecté par le traitement.
Une épiphysiolyse de la tête fémorale peut être observée après l'arrêt du traitement par la GnRHa. Il semblerait que les faibles taux d'œstrogènes induits au cours du traitement par des agonistes de la GnRH affaibliraient la plaque épiphysaire. L'augmentation de la vitesse de croissance à l'arrêt du traitement entraîne alors une diminution des forces de cisaillement nécessaires pour déplacer l'épiphyse.
INTERACTIONS |
Connectez-vous pour accéder à ce contenu
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Grossesse
ZEULIDE 3,75 mg est contre-indiqué chez les femmes qui sont enceintes ou pourraient devenir enceintes alors qu'elles reçoivent le médicament (voir rubrique Contre-indications).
L'injection d'acétate de leuproréline peut avoir des effets néfastes sur le fœtus lorsqu'elle est administrée pendant la grossesse. Toute grossesse doit être exclue avant de débuter le traitement par ZEULIDE 3,75 mg. Un avortement spontané peut se produire si le médicament est administré pendant la grossesse.
Allaitement
On ignore si l'acétate de leuproréline est excrété dans le lait maternel. Par conséquent, ZEULIDE 3,75 mg ne doit pas être utilisé chez les femmes allaitantes. Voir rubrique Contre-indications.
Fertilité
Les études menées chez l'animal ont mis en évidence que la leuproréline pouvait avoir des effets sur la fertilité (voir rubrique Sécurité préclinique).
CONDUITE et UTILISATION DE MACHINES |
L'aptitude à conduire des véhicules et à utiliser des machines peut être altérée par des troubles visuels et des vertiges.
EFFETS INDÉSIRABLES |
Connectez-vous pour accéder à ce contenu
SURDOSAGE |
Il n'y a aucune expérience clinique des effets d'un surdosage significatif de ZEULIDE 3,75 mg ou d'acétate de leuproréline. Lors d'essais cliniques dans lesquels de l'acétate de leuproréline était administré quotidiennement en injection sous-cutanée chez des patients atteints d'un cancer de la prostate, des doses atteignant 20 mg/jour pendant deux ans n'ont causé aucun effet indésirable en dehors de ceux observés avec une dose de 1 mg/jour.
Dans des études menées sur des animaux, des doses allant jusqu'à 500 fois la dose humaine recommandée ont provoqué une dyspnée, une baisse de l'activité et une irritation locale au point d'injection. En cas de surdosage, le patient doit être étroitement surveillé et sera traité de façon symptomatique.
PHARMACODYNAMIE |
Connectez-vous pour accéder à ce contenu
PHARMACOCINÉTIQUE |
Connectez-vous pour accéder à ce contenu
SÉCURITÉ PRÉCLINIQUE |
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogenèse, et des fonctions de reproduction et de développement conduites avec l'acétate de leuproréline, n'ont pas révélé de risque particulier pour l'homme.
Comme attendu et au vu des propriétés pharmacologiques connues, les études non cliniques ont montré des effets réversibles sur le système de reproduction. Dans les études de toxicité pour la reproduction, l'acétate de leuproréline n'a présenté aucune tératogénicité. Cependant, une embryotoxicité/létalité a été observée chez le lapin.
Des études de cancérogénicité réalisées chez le rat avec l'acétate de leuproréline administré par voie sous-cutanée (0,6 à 4 mg/kg/jour), ont montré une augmentation dose-dépendante des adénomes hypophysaires. De plus, une augmentation significative mais non dose-dépendante des adénomes langerhansiens du pancréas chez la femelle et des adénomes des cellules interstitielles testiculaires chez le mâle a été observée, en regardant l'incidence la plus élevée dans le groupe recevant de faibles doses. L'administration d'acétate de leuproréline a entraîné l'inhibition de la croissance de certaines tumeurs hormonodépendantes (tumeurs prostatiques chez des rats Noble et Dunning et tumeurs mammaires induites par le DMBA chez des rats femelles). Aucun effet semblable n'a été observé dans les études de cancérogénicité réalisées chez la souris. Aucune étude de cancérogénicité n'a été réalisée avec ZEULIDE 3,75 mg.
L'acétate de leuproréline ne s'est pas révélé mutagène lors de tests réalisés in vitro et in vivo. Aucune étude de mutagénicité n'a été réalisée avec ZEULIDE 3,75 mg.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Aucun solvant autre que le solvant stérile fourni pour ZEULIDE 3,75 mg ne peut être utilisé pour la reconstitution de la poudre de ZEULIDE 3,75 mg.
DURÉE DE CONSERVATION |
3 ans.
Une fois reconstitué avec le solvant, la suspension doit être administrée immédiatement.
PRÉCAUTIONS PARTICULIÈRES DE CONSERVATION |
A conserver à une température ne dépassant pas 25 °C. Ne pas congeler.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
PRÉCAUTIONS PARTICULIÈRES D'ÉLIMINATION ET DE MANIPULATION |
Mode d'administration
Le flacon de ZEULIDE 3,75 mg doit être reconstitué immédiatement avant l'administration du médicament par une unique injection intramusculaire. Assurez-vous d'employer une technique aseptique.
La solution reconstituée est une suspension d'aspect laiteux, de couleur blanche.
Utilisez le solvant inclus dans le kit du produit. Aucun autre solvant ne peut être utilisé pour la reconstitution de ZEULIDE 3,75 mg.
Ce médicament est destiné à une seule injection. Toute solution restante non utilisée doit être éliminée.
Reconstituer ZEULIDE 3,75 mg conformément aux instructions suivantes. Lisez attentivement avant d'administrer le produit :
| 1 |
|
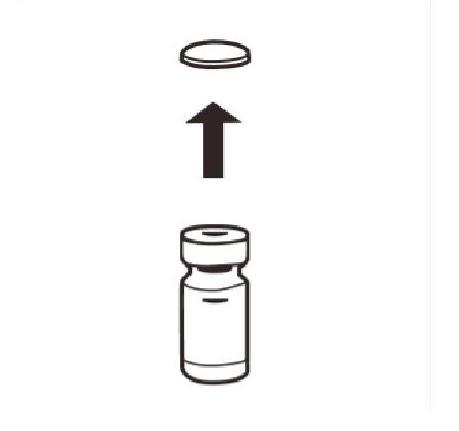
Retirer complètement la capsule du dessus du flacon pour faire apparaître le bouchon en caoutchouc. Vérifier qu'aucun morceau de la capsule ne reste sur le flacon. |
|
| 2 |
|
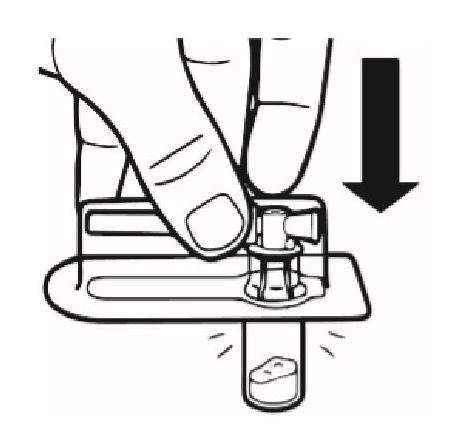
Placer le flacon verticalement sur une table. Retirer l'opercule de la plaquette contenant l'adaptateur pour flacon (MIXJECT). Ne pas retirer l'adaptateur pour flacon de la plaquette. Placer fermement la plaquette contenant l'adaptateur pour flacon sur le dessus du flacon et percer le flacon en position totalement verticale. Appuyer délicatement jusqu'à sentir que l'adaptateur est enclenché. |
|
| 3 |
|
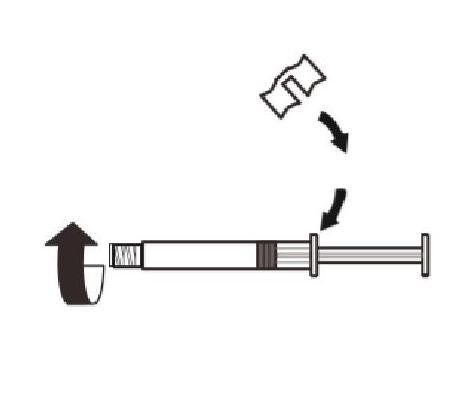
Apposez l'agrippoir blanc sur la seringue jusqu'à ce qu'il s'enclenche. |
|
| 4 |
|
Attachez la seringue à l'adaptateur du flacon en la vissant dans le sens des aiguilles d'une montre dans l'ouverture sur le côté de l'adaptateur du flacon. |
|
| 5 |
|
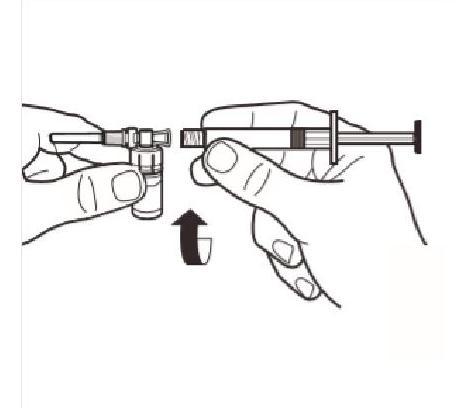
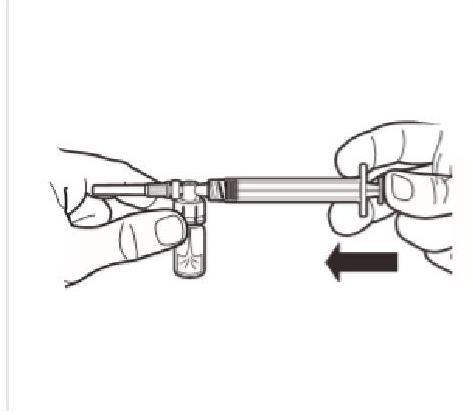
Tout en maintenant la seringue et le flacon bien attachés en position droite, appuyez doucement sur le piston pour transvaser tout le diluant dans le flacon. |
|
| 6 |
|
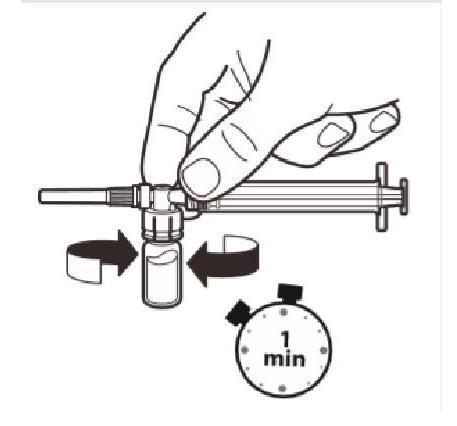
La seringue toujours raccordée au flacon, secouez doucement le flacon pendant environ une minute jusqu'à obtenir une suspension uniforme d'un blanc laiteux. |
|
| 7 |
|
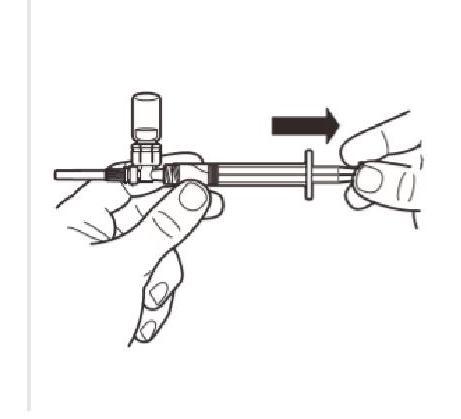
Retournez le système MIXJECT de manière à ce que le flacon soit en haut. Saisissez fermement le système MIXJECT par la seringue et tirez doucement sur la tige du piston pour prélever le produit reconstitué dans la seringue. |
|
| 8 |
|
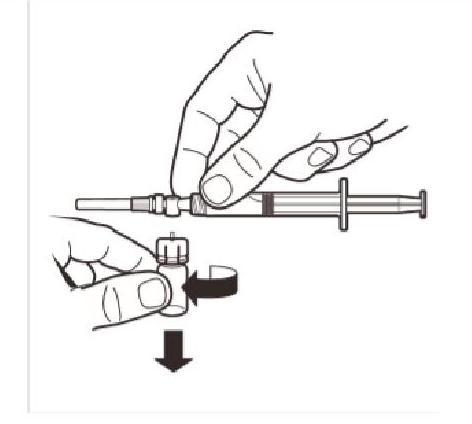
Détachez l'adaptateur du flacon de l'assemblage MIXJECT-seringue : Saisissez fermement la seringue et tournez le flacon (en tenant le capuchon en plastique de l'adaptateur) dans le sens des aiguilles d'une montre. |
|
| 9 |
|
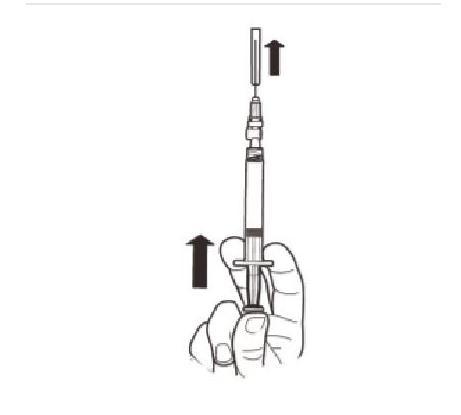
Tenez la seringue DROITE. De l'autre main, tirez le capuchon de l'aiguille vers le haut. Appuyez sur le piston pour évacuer l'air de la seringue. La seringue contenant le produit est prête pour une administration immédiate. |
|
| 10 |
|
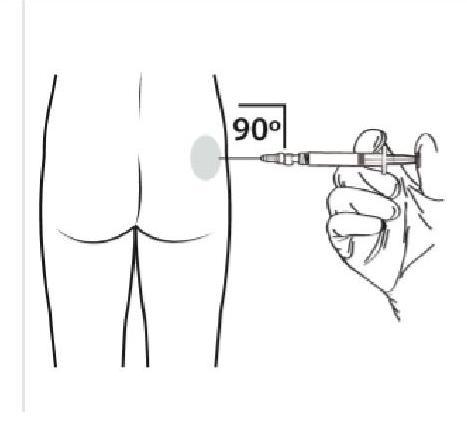
Administrez l'injection intramusculaire en insérant l'aiguille selon un angle de 90 degrés dans la région glutéale. Veillez à ce que la quantité totale du produit soit injectée. Il faut alterner les points d'injection. |
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM |
|
| Prix : |
|